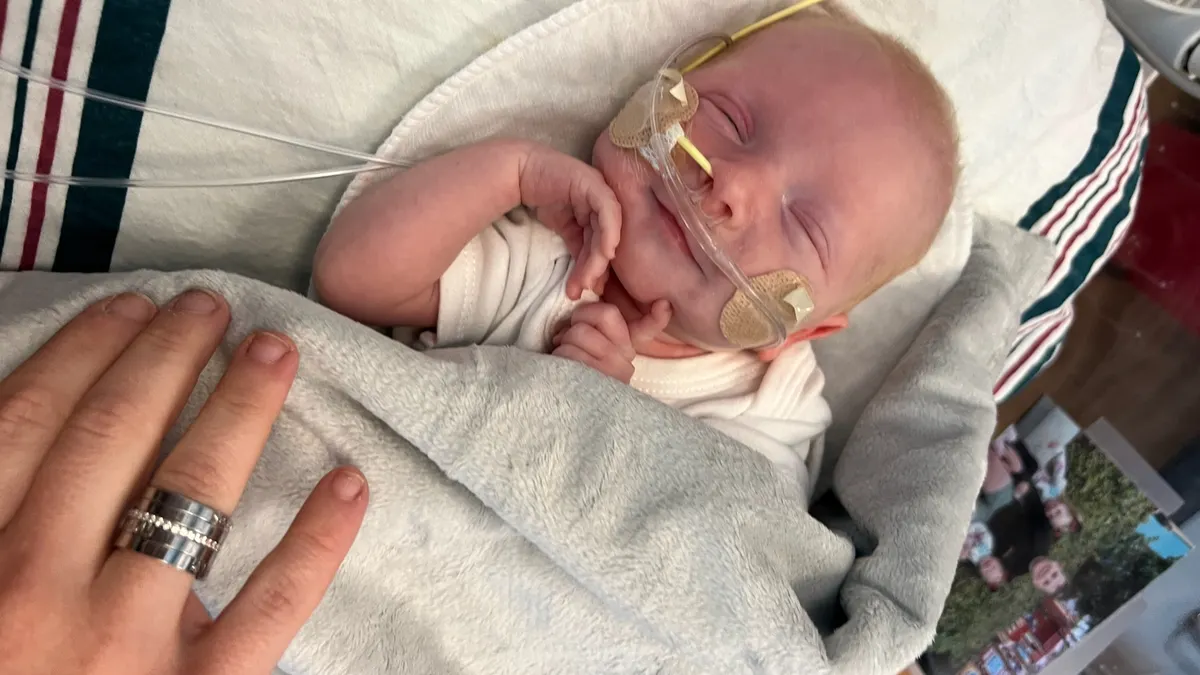
A group of scientists successfully made a bespoke gene editing medicine for a critically ill baby in just a few months, suggesting CRISPR technology could be used to quickly develop personalized therapies for an array of ultra-rare diseases.
Study results published in The New England Journal of Medicine and presented at a medical meeting Thursday reveal that a treatment tailored to an infant with a deadly metabolic disorder was safely administered. They also describe early evidence the treatment has helped stabilize the infant’s disease.
Researchers at the Children’s Hospital of Philadelphia and several other institutions designed and developed the treatment within seven months of the baby’s birth. It’s meant to correct a specific genetic abnormality that causes the metabolic disorder, known as CPS1 deficiency for short.
Part of a family of “urea cycle disorders” that disrupt liver metabolism, CSP1 deficiency results in ammonia accumulation that’s toxic to the brain. Treatment, while limited in effect, typically involves diet restrictions, dialysis and certain drugs.
After receiving three doses of the therapy, the baby, named KJ and now nearly 10 months old, can consume more protein and requires less supportive medication. KJ has also withstood multiple viral infections that might normally worsen his condition.
“All the milestones that he's reaching, or the developmental moments that he's reaching, show us that things are working,” Nicole Muldoon, his mother, told reporters in a media briefing this week.
Longer follow-up is needed to determine how much the therapy actually ameliorates KJ’s disease and improves his long-term health. Doctors also couldn’t yet safely perform the liver biopsy that’s needed to show the treatment’s effects on a genetic level, leaving important questions unanswered.
“We are still in very early days,” said Rebecca Ahrens-Nicklas, an assistant professor of pediatrics at the University of Pennsylvania and study author. Doctors will monitor KJ’s progress, and are considering other ways to evaluate the therapy’s effects without a biopsy.
Yet the findings could carry important implications for drug research. There are more than 7,000 rare diseases, many of which are so uncommon they’re unlikely to be profitable for any companies that develop treatments for them. Gene editing could be a powerful solution, but an expensive development path and slim sales prospects make such medicines tough investment propositions. A large number of biotechnology firms pursuing gene editing are struggling to survive.
Speeding development of gene editing therapies tailored to individuals may be one answer. In an editorial also published in NEJM Thursday, Peter Marks, the former head of the Food and Drug Administration office that regulates gene editing, wrote that a “forward leaning, science-based regulatory approach” might address the commercial challenges limiting this approach’s use against these so-called N-of-1 disorders.
The results published Thursday, while “very early,” are a clear example, he wrote.
KJ’s case adds to other instances in recent years of researchers designing custom therapies for specific individuals. In 2018, a girl named Mila with Batten disease was given a bespoke medicine made using an older drugmaking technology. Scientists at Boston Children’s Hospital have followed that model several times since.
KJ’s disease is extremely uncommon, affecting an estimated one in every 1.3 million people born. While the condition’s severity can vary, its most serious form takes hold in early infancy and causes a panoply of potentially life-threatening health problems. Liver transplants may help, but babies diagnosed with the disease can suffer irreversible brain damage before they’ve grown enough to receive one. More than half die, according to Ahrens-Nicklas.
KJ experienced high ammonia levels immediately after birth and was quickly put on dialysis. He received supportive care at CHOP, and was closely monitored for serious health problems. The typical plan in cases like his was to wait until he could receive a new liver, she said.
Ahrens-Nicklaus had a different idea, though. For a few years, she had worked closely with Kiran Musunuru, a professor of medicine at UPenn who specializes in CRISPR gene editing. The two were collaborating on ways to develop personalized, gene editing therapies that could turn on enzymes that were defective or missing in people like KJ.
By the time KJ was born, they had already completed multiple “practice runs” of a monthslong process to design and test a corrective therapy, Musunuru said.
Once KJ was diagnosed, “we were ready,” he added.
Within four months, the researchers sketched out what they believed would be the most effective therapy design and met with the FDA to discuss testing it on KJ. They conducted preclinical safety tests over the next two months, and constructed a batch of the drug. The FDA cleared their clinical application one week after filing, enabling Ahrens-Nicklaus, Musunuru and the rest of the team to dose KJ when he was 7 months and 8 months old. KJ recently received third dose of the drug, which is expected to be his final infusion.
“The FDA recognized that this was an unusual circumstance. KJ was very, very sick, and there wasn't time for business as usual,” Musunuru said. “They told us to do as good a job as we possibly could in the limited time we had, and they would take it from there.”
The speedy process represents a “milestone in the evolution of personalized therapies for rare and ultrarare inborn errors of metabolism,” wrote Alexis Komor, an associate professor at University of California, San Diego and Andrea Gropman, a pediatric neurogeneticist at St. Jude’s Children Hospital, in an accompanying editorial.
Still, they noted the “evidentiary” limitations of such N-of-1 experiments that make it harder to gauge whether the treatment is safe, works or provides lasting benefits.
The findings, Komor and Gropman wrote, “offer hope and yet require validation.”
This type of approach can hold hard-to-predict risks. A bespoke gene therapy designed for a person with Duchenne muscular dystrophy wasn’t successful, and the individual died days after treatment. In a “preprint” paper that hasn’t yet been peer-reviewed, the researchers behind that experiment note how its N-of-1 nature made it more difficult to draw conclusions about the cause of the patient’s death.
Marks, who while at the FDA pushed the agency to be more flexible in reviewing rare disease drugs, is still confident in a path forward, however. The study published Thursday, he wrote, points to a future in which U.S. regulators could “markedly reduce the complexity and cost of product development.”
Perhaps a gene editing drug for a rare disease could receive an initial approval for its “overall approach,” he said. If so, a slight switch of a single component, like the guiding sequence of RNA, could allow its use in similar-but-different diseases. That “could transform N-of-1 therapy into N-of-many therapies, thus leading to commercial viability of these products for rare diseases,” Marks wrote.
“I don't think I'm exaggerating when I say this is the future of medicine,” Musunuru said. “We very much hope we are showing it's possible to make a personalized gene editing therapy for a single patient, in real time in several months, and it will inspire others to do the same.”