Almost a year ago, a barely six-month-old baby became the first person to receive a personalized therapy made with CRISPR technology.
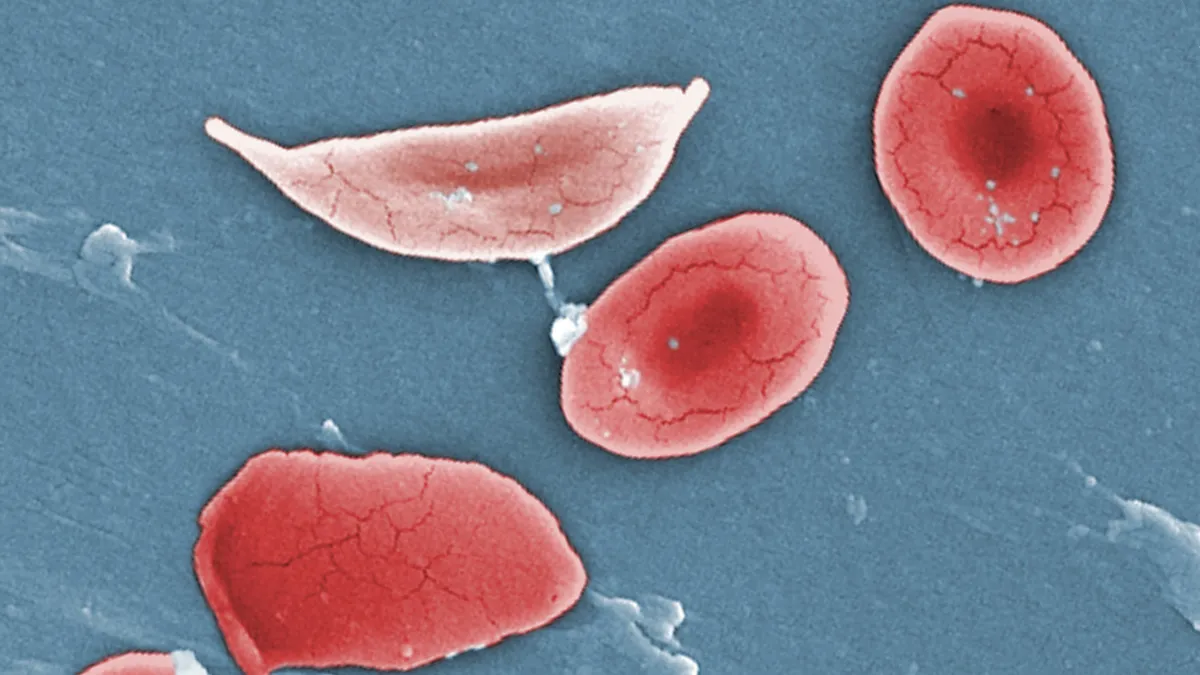
KJ Muldoon was born with an extremely rare, frequently fatal disease where genetic mutations turn blood toxic. He’s alive today thanks to researchers at the Children’s Hospital of Philadelphia and several other institutions, who were able to rapidly design and manufacture a bespoke, gene-editing treatment that’s so far helped stabilize his condition.
Robert F. Kennedy Jr., secretary of the Department of Health and Human Services, said that after hearing about KJ’s story, he directed his agency to make sure this “breakthrough technology would not remain a one-time medical miracle.” Those efforts culminated Monday, with the release of new guidelines for drug developers hoping to create custom treatments for patients with extremely uncommon diseases.
“For ultra-rare conditions, randomized control trials are often — and mostly always — just not feasible,” Kennedy said. “Under the framework that we're announcing today, one well-controlled clinical investigation, supported by confirmatory evidence, can support approval.”
“For decades, families heard the same thing: there are not enough patients, the approval will take too long, you just have to wait [for] the science to catch up with your child. That ends today,” he added.
The new draft guidance from the Food and Drug Administration offers a more detailed look at the “plausible mechanism pathway” that agency leaders Martin Markary and Vinay Prasad first described late last year, in an article published by The New England Journal of Medicine. That pathway is meant to spur the development of therapies for diseases so rare they make little economic sense for drugmakers.
The FDA notes how, just like with other drugs, individualized therapies must be backed by “substantial evidence” showing they’re effective and safe when used as intended.
A developer planning to use this system should: provide a clear connection between a specific genetic abnormality and a disease; demonstrate its therapy takes aim at either the root cause of a disease or a related biological pathway; rely on “well-characterized” natural history data in untreated patients; and be able to confirm its therapy can successfully drug or edit the target.
Additionally, the FDA recommends that, for these therapies to receive traditional approval, they need to improve clinical outcomes, the course of patients’ disease, or biological markers that predict clinical benefit.
Trials for ultra-rare diseases can be “really hard to run,” according to Rebecca Ahrens-Nicklas, an attending physician at CHOP who was on Muldoon’s treatment team and also serves as associate chief for research for the hospital’s human genetics division.
“Add regulatory uncertainty, high per-patient development costs and uneven access to diagnostic testing and expert clinical care — it is really easy to see why promising science too often stalls before it reaches patients,” she said. “This is why clear, pragmatic, consistent regulatory guidance is essential. The types of individualized genetic therapies that we're trying to develop simply do not fit in the traditional model of drug development.”
While the fresh guidance specifically discusses genome editing and RNA-based therapies, the FDA stressed that it’s applicable to other drugmaking methods as well. Broadly, the hope is this framework will allow for one therapy to be approved for, say, a few mutations in a particular disease, and then enable other similar therapies to soon follow.
Leaders of HHS and FDA said it could even be used for more common diseases that fit the criteria of debilitating or life-threatening and particularly difficult to evaluate through a randomized, controlled clinical trial.
Tracy Beth Høeg, acting director of the FDA’s main drug review arm, called the guidance “transformational,” expecting it to ignite the biopharmaceutical industry’s interest in personalized therapies. The agency is “likely going to be overwhelmed with applications for these treatments for these rare diseases,” she said.
One biotechnology company, Aurora Therapeutics, has already emerged explicitly to take advantage of the plausible mechanism pathway. Aurora was co-founded by Nobel laureate Jennifer Doudna and genetic medicine expert Fyodor Urnov, and is helmed by Ed Kaye, the former leader of Sarepta Therapeutics and Stoke Therapeutics.
“We don't want investors scared away because of the high cost,” Makary said. “We want investors to see the bright promise of rare disease therapies.”
The FDA estimates more than 30 million people in the U.S. have a rare disease, which is defined as a condition affecting less than 200,000 people in the country.