
A decade ago, scientists outlined the gene editing potential of CRISPR, turning the vestiges of a bacterial immune system into one of the biotechnology industry’s most powerful tools.
On Tuesday, a group of advisers to the Food and Drug Administration met to discuss the merits of what could be the first CRISPR medicine approved by the agency: a treatment for sickle cell disease from partners Vertex Pharmaceuticals and CRISPR Therapeutics.
Documents published last week show that FDA scientists are focused on the technical aspects of how CRISPR does its DNA-editing work. They seem relatively convinced the treatment, known as exa-cel, is effective.
BioPharma Dive tracked the daylong meeting and reported on the discussion here. The most recent entries are listed first.
4:15 pm
About an hour before the meeting was scheduled to end, OTP Director Verdun thanked public speakers, Vertex and FDA staff as she drew the day’s discussions to a close.
“An important part of our mission is not just evaluating efficacy, but safety, both short- and long-term, and doing what we can to evaluate both known and unknown risk of therapies,” Verdun said.
She noted that the FDA will take the committee’s discussions and recommendations into consideration as it completes its review of exa-cel.
While the FDA doesn’t have to follow the advice of its advisory panels, it often does. The agency is set to decide on exa-cel’s approval by Dec. 8. — Gwendolyn Wu
4:00 pm
Committee chair Ahsan closed the discussion by summarizing the panelists’ views. They agreed, she said, that Vertex’s analysis of off-target risks was reasonably detailed, but indicated further study could still be useful. “So then the question becomes, when have we done enough theoretical analysis to allow us to move forward?” she said.
Ahsan added that, to better understand if and how off-target edits emerge, Vertex needs to continue to monitor patients. Yet she acknowledged that approach might have some limitations, however.
“It would be nice to see some evaluation of monitoring the edits over real time, looking at clonal expansion,” Ahsan said. “But it's unsure the technology that would be used to do that, whether whole genome sequencing [or something else], would actually have the detection levels to give us meaningful information there.” — Jonathan Gardner
3:40 p.m.
The committee has ended its cross-examination of Vertex and moved on to the broader discussion. Scot Wolfe, professor of molecular, cell, and cancer biology at the University of Massachusetts medical school, summarized Vertex’s methods as “pretty detailed,” and quibbled only with the depth of the analysis.
“We want to be careful to not let the perfect be the enemy of the good,” Wolfe said. “You want to do as good a job as you possibly can, but at some point, you have to try things in patients. I think in this case, there is a huge unmet need.”
That was echoed by Alexis Komor, an assistant professor of chemistry and biochemistry at University of California, San Diego. “Do we have the technology to sequence every single patient and do an expansive individualized on-target analysis on each one? Probably, but is that reasonable to expect from them at this point? I don’t know,” she said. — Jonathan Gardner
3:30 p.m.
FDA panelists asked a number of questions to agency reviewers and to Vertex. Joseph Wu, one of the panelists and director of the Stanford Cardiovascular Institute, noted how Vertex’s cellular off-target analysis used donor cells from only three sickle cell patients, while the company has treated dozens more in the exa-cel trial.
“You've had several years with these samples. Why not just do the actual analysis rather than the in silico modeling?" Wu said.
The sickle cell patient samples Vertex used were among 14 donor cell samples that the company has tested, and “there's no data to suggest that the result would be different for patients with sickle cell than the other possibilities,” Altshuler responded.
“Whether we were to do the testing,” Altshuler said, “we'd end up at the same place, I believe, which is the risk assessment.”
Wu’s line of questioning, as well as the concerns raised by Singh in her analysis, return to a similar theme: how much off-target analysis is enough?
“I suspect if there were hard guidelines we wouldn't be having this meeting,” committee chair Ahsan said. — Gwendolyn Wu
3:05 p.m.
FDA staff just finished outlining the agency’s view of exa-cel. The first presentation, by reviewer Karl Kasamon, gave a clinical overview of the therapy and largely lined up with Vertex’s assessment of its efficacy. The second, by bioinformatics expert Komudi Singh, was a detailed review of the day’s main issue: off-target CRISPR editing.
Singh described how off-target edits, while potentially inconsequential, can be harmful if they occur in regions of the genome that have regulatory functions or code for a protein. There are a number of tools available to assess this likelihood and Vertex took several approaches, including using computer algorithms to identify possible off-target sites and sequencing donor cells that were edited.
Singh raised a number of concerns with Vertex’s methods. For example, the database used by the company contains only a small amount of sequencing data from the intended population for exa-cel treatment. It also may not be representative of genetic variants that present higher risk of off-target edits. Vertex’s cellular analysis, meanwhile, used a small sample of donor cells.
Singh ended by asking the committee to provide recommendations, setting up the day’s concluding discussion on whether other studies are needed to gauge exa-cel’s safety. — Ned Pagliarulo
1:12 p.m.
The open public hearing began with Victoria Gray, a 38-year-old woman who was the first patient to be treated with exa-cel. She spoke of sickle cell’s pain, which she compared to being simultaneously hit by a truck and lightning, and the disease’s effects on her life and family.
She met with physician Haydar Frangoul, who offered her a spot in the exa-cel trial. “I said yes without hesitation, knowing that I would be the first person but this was my opportunity to fight,” Gray recounted.
She no longer has pain crises after receiving exa-cel, nor does she need blood transfusions. “I now work full time and I contribute to my household and my community.”
Her experience was similar to that of Jimi Olaghere, who participated in the exa-cel study about three years ago.
“Gene therapy has given me the ability to take full control of my life,” said Olaghere. “In a world where the deck was stacked against me, gene therapy has been a winning hand. While I recognize gene editing won’t be the solution for everyone I strongly recommend [sickle cell] warriors to consider this one-time therapy.” — Ned Pagliarulo
12:10 p.m.
The panel is now on break until 12:35 p.m., when we'll hear from patients and advocates during the open public hearing. — Ned Pagliarulo
12:05 p.m.
The first two patients who entered the exa-cel trial have seen notable improvements since they were treated, said Haydar Frangoul, hematology and oncology medical director at Tristar Centennial Medical Group in Nashville and lead investigator of the exa-cel studies.
One, a 33-year-old woman who “couldn't walk or even pull up a spoon to feed herself” during pain crises, has been able to take up full-time work and become more active in caring for four children since receiving the therapy. Another, a 13-year-old girl who was hospitalized several times each year because of pain crises, can attend school regularly because she no longer needs as much medical care.
Frangoul also urged exa-cel’s use early in the disease course because of the cumulative effects of the disease on organs and joints.
“Sickle cell disease is like a hammer hitting a wall,” he said. “I can take away the hammer. But we cannot reverse the damage. We cannot fix the wall.” — Jonathan Gardner
11:45 a.m.
To track exa-cel safety over the long term, Vertex plans to rely on two 15-year studies, including a registry-based study that would begin post-approval and involve sickle cell patients treated with exa-cel. The other is follow-up for an ongoing clinical trial.
The Center for International Blood and Marrow Transplant Research, which has for years collected data on patients receiving cell therapies, will also assess the long-term safety of exa-cel, said Christopher Simard, Vertex's vice president of global patient safety. All planned U.S. exa-cel treatment centers will provide data to the center, he said. — Gwendolyn Wu
11:10 a.m.
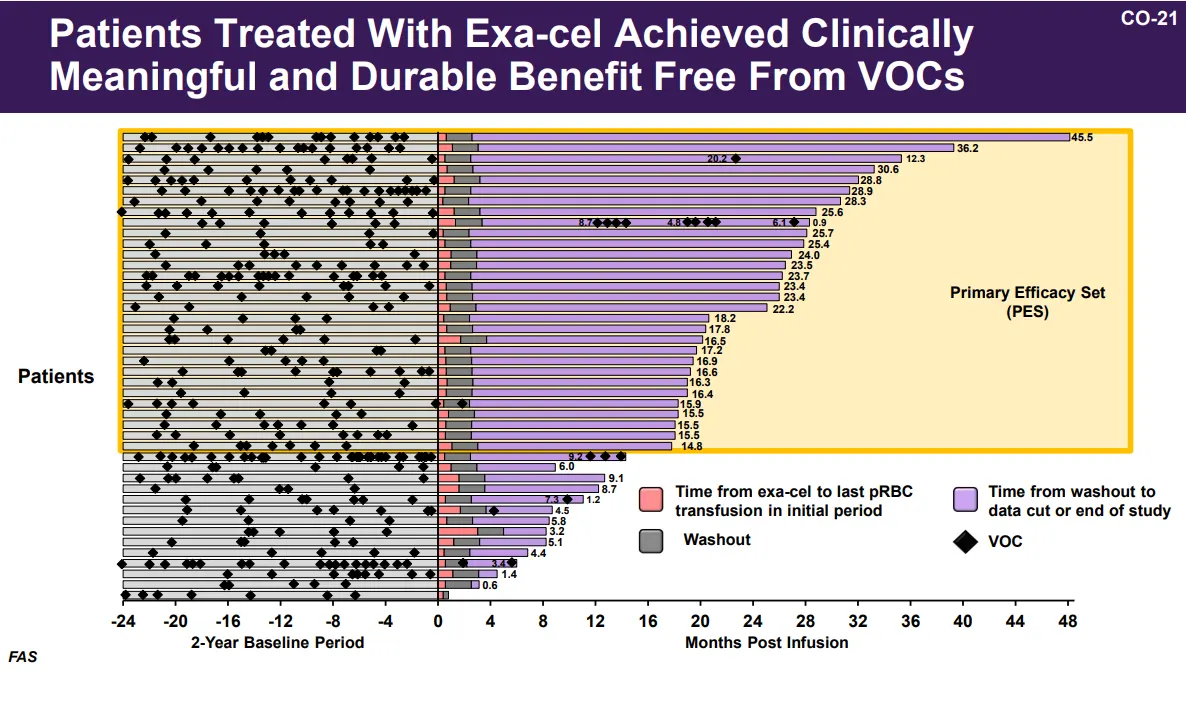
William Hobbs, Vertex’s head of hematology clinical development, outlined the clinical trial data showing exa-cel’s effectiveness. The results are clear: Treatment eliminates, for at least multiple years, the debilitating pain crises that people with sickle cell can experience and keeps them out of the hospital. — Ned Pagliarulo
11:05 a.m.
There are a few drugs currently available to help manage sickle cell symptoms. But they don’t fix the condition and don’t work for everyone.
A bone marrow transplant of donor stem cells can cure the disease, but is available for less than 20% of people with sickle cell, said Alexis Thompson, division chief of hematology at Children’s Hospital of Philadelphia, who spoke at Tuesday’s meeting on behalf of Vertex.
An estimated 100,000 people live with sickle cell in the U.S., and about one-fifth have a severe form of the disease, with recurring symptoms such as severe pain flare-ups, a lung condition known as acute chest syndrome, priapism and splenic sequestration. The resulting organ damage raises the risk of death.
Sickle cell also occurs at "disproportionately high rates" among people of color, particularly people of African ancestry, Thompson said.
“People with sickle cell often live in low income areas and communities with high unmet medical need, further adding to substantial healthcare disparities,” she said. — Gwendolyn Wu
10:50 a.m.
The vastness of the human genome presents a thorny problem for assessing the risk of off-target CRISPR edits. Scientists trying to vet the risk have to focus their analysis, or risk being swamped by a deluge of data that might not actually help suss out potential problems.
Urnov, answering a question from the panelist and National Institutes of Health branch chief John Tisdale, said there’s a limit to how much reassurance preclinical editing analyses can provide.
“The technology is in fact ready for prime time,” said Urnov. “We're kind of reaching asymptotic places in terms of how we can de-risk it non-clinically. I don't know what else to do at this point in terms of understanding the benefit-risk.”
One advantage to studying blood diseases, Bauer noted, is that samples can be easily obtained, tested and tracked over time.
Taby Ahsan, the committee’s chair, said Urnov’s and Bauer’s answers set the stage for the rest of the day’s discussion: “When is enough theoretical data sufficient to support a patient specific risk assessment? And where are we in that curve of risk mitigation?” — Ned Pagliarulo
10:25 a.m.
After deeply technical presentations by Urnov and Daniel Bauer about the risks of off-target edits, committee member Lisa Lee, a bioethics expert from Virginia Tech University, brought it back to the patient level by asking a simple question: “If you were talking to a family about this kind of treatment, how would you characterize the consequences of off target edits?”
Bauer acknowledged the uncertainty of the risk by noting how most of what’s in the human genome doesn’t code for any specific function, meaning an off-target edit might have no effect on patients.
“The only way to know that is through careful follow up,” he said. “My guess is it's a relatively small risk in the scheme of the risk-benefit. And that's one of the goals, I would say, of doing this under very careful circumstances is to try to learn what that risk is so that we can continually improve those therapies.” — Jonathan Gardner
9:50 a.m.
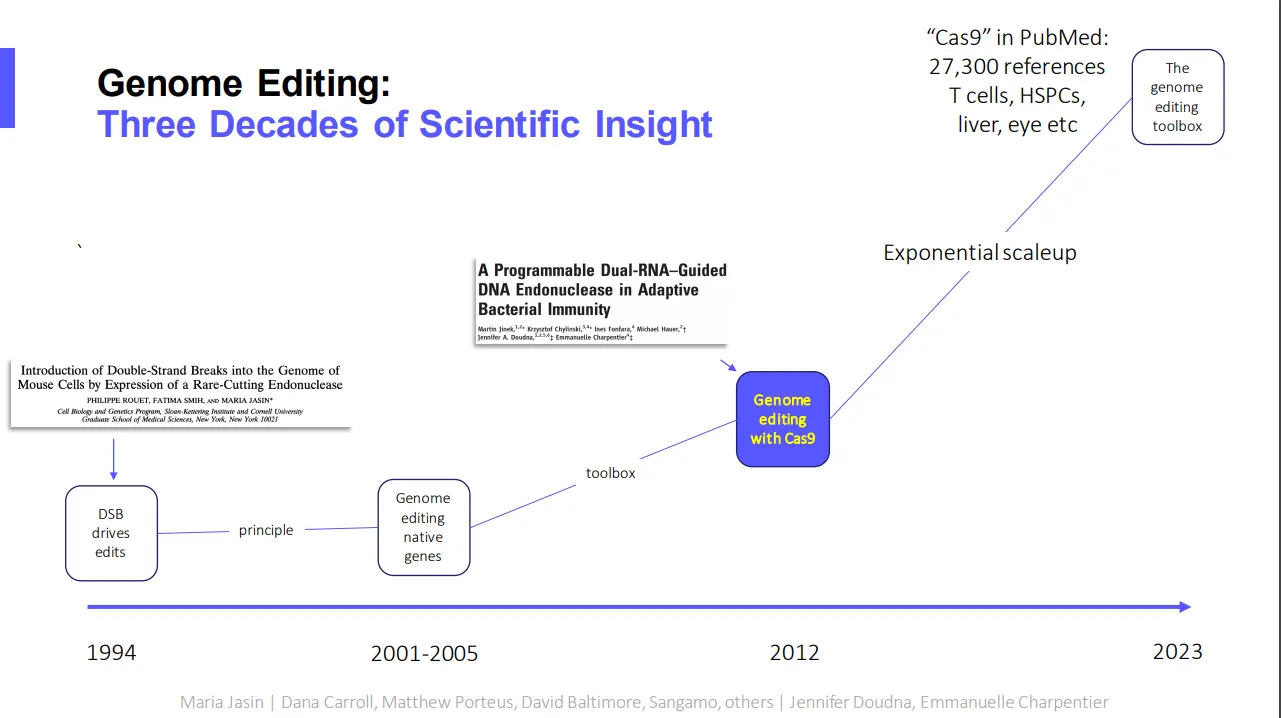
Fyodor Urnov is a leading expert on CRISPR gene editing and director of technology and translation at UC Berkeley’s Innovative Genomics Institute. His task today: Condense the history of CRISPR gene editing research to a 20-minute presentation.
He starts by noting that, while CRISPR is still relatively new, it builds on decades of research into gene editing by other means.
“I need to frame the state of our field of gene editing today by stepping 20 years back,” Urnov said. “So at the time, the sole method for targeted genetic engineering in human cells was an approach called gene targeting.” It was inefficient and toxic, he added, ruling out therapeutic applications.
The discovery of CRISPR has brought about an “exponential scale-up” in gene editing research and its potential applications — a shift Urnov documented with the stylized chart below.
“Here we are in 2023, and we are proverbially in a whole new world. There are 27,000 references to the word cas9 in PubMed. Genome editing with cas9 and other tools has been shown to work in every basic and applied research setting where it can be tried, as well as in clinical trials of blood stem cells, T cells, the liver and the eye. — Ned Pagliarulo
9:30 a.m.
OTP director Verdun kicked off the meeting by emphasizing the FDA convened the meeting to specifically evaluate the risk of off-target edits. Her comments again signaled that agency scientists are comfortable exa-cel helps patients and that its safety profile is otherwise acceptable.
Verdun also added a personal note about her experience with sickle-cell patients.
“I've had the pleasure of taking care of several sickle cell patients and admire the courageous and resilient patient community,” she said. “I'm also reminded of the sickle cell disease patient-focused drug development program at FDA in which we heard directly from patients and their caregivers which highlighted the significant unmet need.” — Jonathan Gardner
9:10 a.m.
After some housekeeping, advisers will hear first from Nicole Verdun, the FDA’s new head of the Office of Therapeutics Products, which oversees gene therapies like exa-cel.
The meeting includes two guest speaker presentations, from gene editing experts Fyodor Urnov, of the University of California, Berkeley, and Daniel Bauer, of Boston Children’s Hospital.
After a short break, Vertex and CRISPR Therapeutics will then walk through their data, followed by an open public hearing. The FDA is up in the afternoon and committee discussion is scheduled from 3:00 p.m. to 4:50 p.m — Ned Pagliarulo
The advisory committee meeting is one of the final steps in the FDA’s review of exa-cel, which the agency expects to complete by Dec. 8. These meetings offer a rare window into the agency’s thinking midway through an approval review, as well as a chance to publicly vet companies’ data.
In this case, the meeting will also serve as a forum for discussing CRISPR gene editing, which has become an important biomedical tool used by a growing number of biotechnology companies. The day’s agenda shows that advisers will hear from other experts about CRISPR’s merits and risks, making Tuesday’s meeting a mini-summit on the technology.
A positive review by the panel would up the chances that the regulator grants the therapy a historic approval, as well as boost other gene editing companies like Intellia Therapeutics. — Ned Pagliarulo
Essentially, exa-cel combines CRISPR gene editing with bone marrow transplantation.
To construct the treatment, a patient’s stem cells are collected from their blood and sent to a manufacturing facility where they are edited using CRISPR/cas9. The DNA snip is made to a specific part of a gene called BCL11A that controls production of a protein known as fetal hemoglobin, or HbF. The edited cells are frozen and shipped back to the treating hospital, where, after a preparatory chemotherapy regimen, they are infused into the patient.
Churning out fetal hemoglobin, the new cells effectively mute the most prominent symptoms of sickle cell, which is caused by damaged adult hemoglobin warping red blood cells into a crescent.
Vertex and CRISPR Therapeutics are seeking approval of exa-cel in people aged 12 years and older who have sickle cell and experience the disease’s characteristic pain crises. — Ned Pagliarulo
Documents published last week showed FDA staff to be laser focused on the risk of “off-target” edits, or when CRISPR makes unintended cuts to DNA other than what it’s been programmed to snip. Wayward edits could disrupt cell functions or cause damage that later leads to health problems.
“Since unintended edits can disrupt gene expression if present in the coding or regulatory DNA sequences, it is critical that the specificity of [exa-cel’s targeting component] be thoroughly screened to ensure off-target genome editing is minimized,” FDA staff wrote.
Vertex and CRISPR have done several analyzes to document and predict this risk with exa-cel specifically, and claim their work shows no detectable off-target editing.
But the FDA appears concerned that their work might have missed something, and wants its advisers to discuss whether any further study is needed. In particular, they raise questions about how well Vertex and CRISPR’s analyses capture the risk of off-target edits in a broad population of people with sickle cell.
On the efficacy side of the equation, however, FDA scientists seemed supportive of exa-cel’s potential, describing Vertex and CRISPR’s data as “strongly positive” at one point in the documents. — Ned Pagliarulo













