By and large, the history of the drug industry has been one of pills and proteins. Biotechnology and pharmaceutical firms have come up with a panoply of variation on those two forms, but most approved medicines can be classified into one category or the other.
A growing class of genetic treatments is now becoming the industry’s third main drugmaking platform. They are built from nucleic acids — strands of small, interfering RNA and sequences of replacement DNA — or on enzymes that cut them. Some are wrapped in specialized viruses, while others are entire cells.
These new treatments can turn off disease-causing genes, or replace defective ones. They offer ways to stop muscular atrophy, curb cancer and correct blood diseases like sickle cell.
More are making their way through testing and reaching patients. The Food and Drug Administration has approved nearly two dozen RNA therapies as well as a dozen gene therapies for inherited diseases. Among that latter group is the world’s first CRISPR medicine, Casgevy.
Alongside that research productivity have come setbacks, however. Cancer cases in studies of sickle cell and hemophilia gene therapies renewed safety concerns, as have patient deaths in a neuromuscular disease trial and a heart disease study. The deaths of two patients treated with a Duchenne muscular dystrophy treatment sparked a standoff last year between the FDA and the treatment’s developer, Sarepta Therapeutics.
The FDA has asked for more data from developers in response. But its priorities are newly in question amid changes in leadership at the offices overseeing cell and gene therapies and mixed messaging about its flexibility in reviewing new rare disease treatments. It’s outlined new ways to speed future approvals, but also flip-flopped on previous guidance in delaying or rejecting multiple programs.
Gene therapy’s market potential is also being tested. Many carry seven-figure price tags, raising affordability concerns. Even when insurers cover treatment, only a few have seen strong adoption.
A bespoke CRISPR therapy suggests a blueprint for treating ‘N-of-1’ diseases
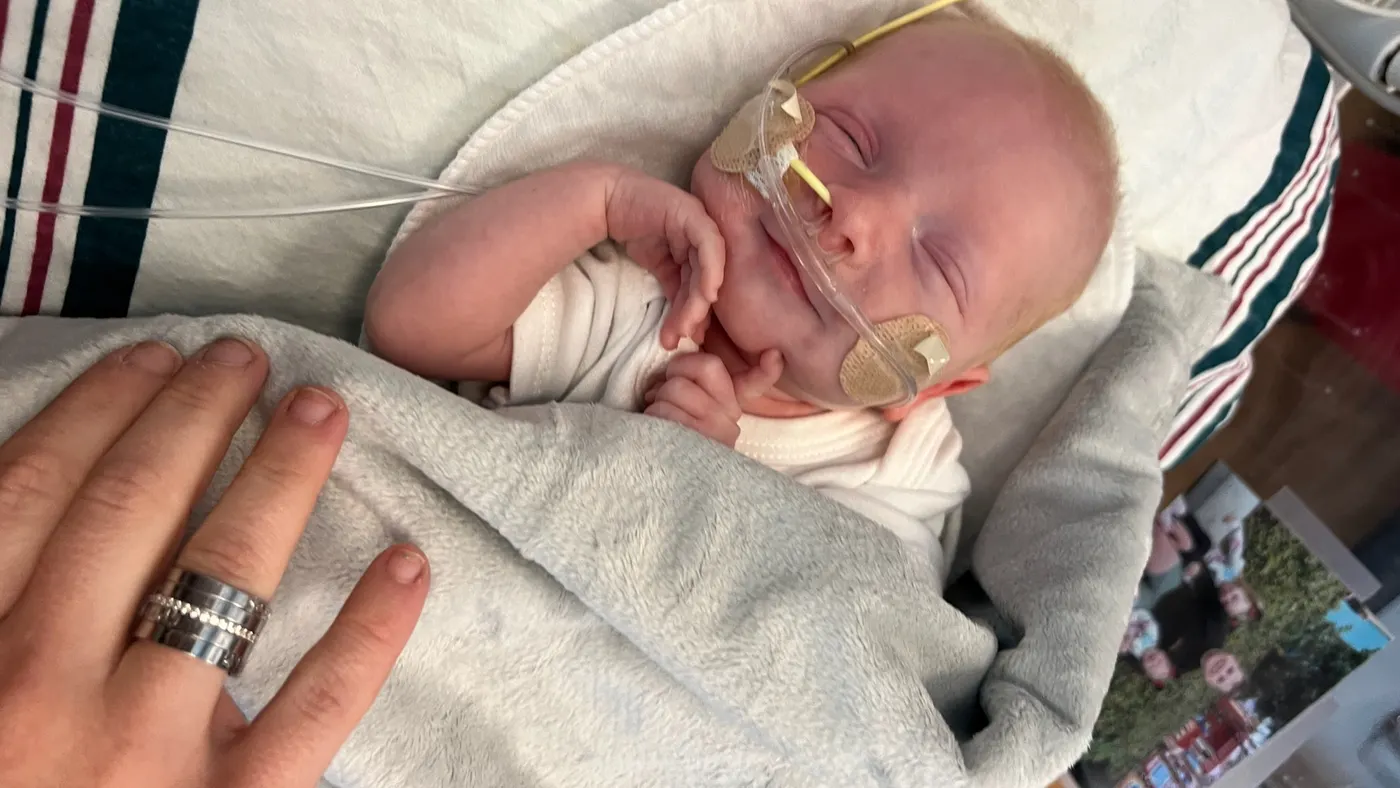
A gene editing drug custom-made for a critically ill baby in just a few months appeared to be safe and working as intended, according to newly published results.
By: Ben Fidler• Published May 15, 2025
A group of scientists successfully made a bespoke gene editing medicine for a critically ill baby in just a few months, suggesting CRISPR technology could be used to quickly develop personalized therapies for an array of ultra-rare diseases.
Study results published in The New England Journal of Medicine and presented at a medical meeting last May revealed that a treatment tailored to an infant with a deadly metabolic disorder was safely administered. They also describe early evidence the treatment has helped stabilize the infant’s disease.
Researchers at the Children’s Hospital of Philadelphia and several other institutions designed and developed the treatment within seven months of the baby’s birth. It’s meant to correct a specific genetic abnormality that causes the metabolic disorder, known as CPS1 deficiency for short.
Part of a family of “urea cycle disorders” that disrupt liver metabolism, CSP1 deficiency results in ammonia accumulation that’s toxic to the brain. Treatment, while limited in effect, typically involves diet restrictions, dialysis and certain drugs.
After receiving three doses of the therapy, the baby, named KJ and about 10 months old in May, can consume more protein and requires less supportive medication. KJ has also withstood multiple viral infections that might normally worsen his condition.
“All the milestones that he's reaching, or the developmental moments that he's reaching, show us that things are working,” Nicole Muldoon, his mother, told reporters in a May 2025 media briefing.
Longer follow-up is needed to determine how much the therapy actually ameliorates KJ’s disease and improves his long-term health. Doctors also couldn’t yet safely perform the liver biopsy that’s needed to show the treatment’s effects on a genetic level, leaving important questions unanswered.
“We are still in very early days,” said Rebecca Ahrens-Nicklas, an assistant professor of pediatrics at the University of Pennsylvania and study author. Doctors will monitor KJ’s progress, and are considering other ways to evaluate the therapy’s effects without a biopsy.
Yet the findings could carry important implications for drug research. There are more than 7,000 rare diseases, many of which are so uncommon they’re unlikely to be profitable for any companies that develop treatments for them. Gene editing could be a powerful solution, but an expensive development path and slim sales prospects make such medicines tough investment propositions. A large number of biotechnology firms pursuing gene editing are struggling to survive.
Speeding development of gene editing therapies tailored to individuals may be one answer. In an editorial also published in NEJM, Peter Marks, the former head of the Food and Drug Administration office that regulates gene editing, wrote that a “forward leaning, science-based regulatory approach” might address the commercial challenges limiting this approach’s use against these so-called N-of-1 disorders.
The results published May 15, while “very early,” are a clear example, he wrote.
KJ’s case adds to other instances in recent years of researchers designing custom therapies for specific individuals. In 2018, a girl named Mila with Batten disease was given a bespoke medicine made using an older drugmaking technology. Scientists at Boston Children’s Hospital have followed that model several times since.
KJ’s disease is extremely uncommon, affecting an estimated one in every 1.3 million people born. While the condition’s severity can vary, its most serious form takes hold in early infancy and causes a panoply of potentially life-threatening health problems. Liver transplants may help, but babies diagnosed with the disease can suffer irreversible brain damage before they’ve grown enough to receive one. More than half die, according to Ahrens-Nicklas.
KJ experienced high ammonia levels immediately after birth and was quickly put on dialysis. He received supportive care at CHOP, and was closely monitored for serious health problems. The typical plan in cases like his was to wait until he could receive a new liver, she said.
Ahrens-Nicklaus had a different idea, though. For a few years, she had worked closely with Kiran Musunuru, a professor of medicine at UPenn who specializes in CRISPR gene editing. The two were collaborating on ways to develop personalized, gene editing therapies that could turn on enzymes that were defective or missing in people like KJ.
By the time KJ was born, they had already completed multiple “practice runs” of a monthslong process to design and test a corrective therapy, Musunuru said.
Once KJ was diagnosed, “we were ready,” he added.
Within four months, the researchers sketched out what they believed would be the most effective therapy design and met with the FDA to discuss testing it on KJ. They conducted preclinical safety tests over the next two months, and constructed a batch of the drug. The FDA cleared their clinical application one week after filing, enabling Ahrens-Nicklaus, Musunuru and the rest of the team to dose KJ when he was 7 months and 8 months old. KJ recently received third dose of the drug, which is expected to be his final infusion.
“The FDA recognized that this was an unusual circumstance. KJ was very, very sick, and there wasn't time for business as usual,” Musunuru said. “They told us to do as good a job as we possibly could in the limited time we had, and they would take it from there.”
The speedy process represents a “milestone in the evolution of personalized therapies for rare and ultrarare inborn errors of metabolism,” wrote Alexis Komor, an associate professor at University of California, San Diego and Andrea Gropman, a pediatric neurogeneticist at St. Jude’s Children Hospital, in an accompanying editorial.
Still, they noted the “evidentiary” limitations of such N-of-1 experiments that make it harder to gauge whether the treatment is safe, works or provides lasting benefits.
The findings, Komor and Gropman wrote, “offer hope and yet require validation.”
This type of approach can hold hard-to-predict risks. A bespoke gene therapy designed for a person with Duchenne muscular dystrophy wasn’t successful, and the individual died days after treatment. In a “preprint” paper that hasn’t yet been peer-reviewed, the researchers behind that experiment note how its N-of-1 nature made it more difficult to draw conclusions about the cause of the patient’s death.
Marks, who while at the FDA pushed the agency to be more flexible in reviewing rare disease drugs, is still confident in a path forward, however. The study published May 15, he wrote, points to a future in which U.S. regulators could “markedly reduce the complexity and cost of product development.”
Perhaps a gene editing drug for a rare disease could receive an initial approval for its “overall approach,” he said. If so, a slight switch of a single component, like the guiding sequence of RNA, could allow its use in similar-but-different diseases. That “could transform N-of-1 therapy into N-of-many therapies, thus leading to commercial viability of these products for rare diseases,” Marks wrote.
“I don't think I'm exaggerating when I say this is the future of medicine,” Musunuru said. “We very much hope we are showing it's possible to make a personalized gene editing therapy for a single patient, in real time in several months, and it will inspire others to do the same.”
Article top image credit: Permission granted by Muldoon family
FDA fleshes out new roadmap for testing personalized therapies
In February, the agency officially unveiled long-awaited draft guidance meant to help speed the development of bespoke treatments for extremely rare diseases.
By: Jacob Bell• Published Feb. 23, 2026
Last year, a barely six-month-old baby became the first person to receive a personalized therapy made with CRISPR technology.
KJ Muldoon was born with an extremely rare, frequently fatal disease where genetic mutations turn blood toxic. He’s alive today thanks to researchers at the Children’s Hospital of Philadelphia and several other institutions, who were able to rapidly design and manufacture a bespoke, gene-editing treatment that’s so far helped stabilize his condition.
Robert F. Kennedy Jr., secretary of the Department of Health and Human Services, said that after hearing about KJ’s story, he directed his agency to make sure this “breakthrough technology would not remain a one-time medical miracle.” Those efforts culminated with the release in February of new guidelines for drug developers hoping to create custom treatments for patients with extremely uncommon diseases.
“For ultra-rare conditions, randomized control trials are often — and mostly always — just not feasible,” Kennedy said. “Under the framework that we're announcing today, one well-controlled clinical investigation, supported by confirmatory evidence, can support approval.”
“For decades, families heard the same thing: there are not enough patients, the approval will take too long, you just have to wait [for] the science to catch up with your child. That ends today,” he added.
The draft guidance from the Food and Drug Administration offers a more detailed look at the “plausible mechanism pathway” that former agency leaders Martin Markary and Vinay Prasad first described late last year, in an article published by The New England Journal of Medicine. That pathway is meant to spur the development of therapies for diseases so rare they make little economic sense for drugmakers.
The FDA notes how, just like with other drugs, individualized therapies must be backed by “substantial evidence” showing they’re effective and safe when used as intended.
A developer planning to use this system should: provide a clear connection between a specific genetic abnormality and a disease; demonstrate its therapy takes aim at either the root cause of a disease or a related biological pathway; rely on “well-characterized” natural history data in untreated patients; and be able to confirm its therapy can successfully drug or edit the target.
Additionally, the FDA recommends that, for these therapies to receive traditional approval, they need to improve clinical outcomes, the course of patients’ disease, or biological markers that predict clinical benefit.
Trials for ultra-rare diseases can be “really hard to run,” according to Rebecca Ahrens-Nicklas, an attending physician at CHOP who was on Muldoon’s treatment team and also serves as associate chief for research for the hospital’s human genetics division.
“Add regulatory uncertainty, high per-patient development costs and uneven access to diagnostic testing and expert clinical care — it is really easy to see why promising science too often stalls before it reaches patients,” she said. “This is why clear, pragmatic, consistent regulatory guidance is essential. The types of individualized genetic therapies that we're trying to develop simply do not fit in the traditional model of drug development.”
While the fresh guidance specifically discusses genome editing and RNA-based therapies, the FDA stressed that it’s applicable to other drugmaking methods as well. Broadly, the hope is this framework will allow for one therapy to be approved for, say, a few mutations in a particular disease, and then enable other similar therapies to soon follow.
Leaders of HHS and FDA said it could even be used for more common diseases that fit the criteria of debilitating or life-threatening and particularly difficult to evaluate through a randomized, controlled clinical trial.
Tracy Beth Høeg, the former acting director of the FDA’s main drug review arm, called the guidance “transformational,” expecting it to ignite the biopharmaceutical industry’s interest in personalized therapies. The agency is “likely going to be overwhelmed with applications for these treatments for these rare diseases,” she said.
“We don't want investors scared away because of the high cost,” Makary said. “We want investors to see the bright promise of rare disease therapies.”
The FDA estimates more than 30 million people in the U.S. have a rare disease, which is defined as a condition affecting less than 200,000 people in the country.
Article top image credit: Anna Moneymaker via Getty Images
Sponsored
The future of advanced therapies: A CDMO perspective on making gene therapy scalable, affordable and ready for patients
Advanced therapies are entering a decisive phase. After a wave of approvals in ultra-rare and rare indications, developers are now pushing toward broader diseases—which demands a step change in manufacturing scalability, cost and reliability. From a CDMO perspective, success will hinge on unifying deep technical innovation with global, inspection-ready capacity and true partnership models that carry programs from discovery through commercial supply.
Scale alone won’t solve the cost-per-dose challenge that limits broader patient access. Traditional AAV manufacturing approaches (transient transfection in HEK293, baculovirus/Sf9 systems and early stable-line models) have delivered important clinical wins, but struggle to achieve mass production at a viable cost. The next decade requires a “both-and” mindset: continue leveraging known, high-precedence methods while deploying new technologies that materially improve productivity, quality and economics.
Two paths are especially promising. First, producer cell lines (PCLs) are maturing and achieving productivity comparable to triple-transfection systems, with AAV particles that meet key quality attributes: comparable genome and capsid titers (via ddPCR and ELISA), intact particle morphology (TEM), appropriate empty/full ratios (AUC, mass photometry), strong infectivity and expression (in vitro and in vivo) and preserved capsid protein structure (CE-SDS, LC-MS/MS). As licensing frameworks solidify and scalability concerns are addressed, PCLs can unlock a step change in dose output and consistency.
Second, process intensification of transient production is yielding tangible gains now. By running perfusion culture of HEK293 cells followed by high-density chemical transfection, 10-fold increase in titer alongside a >50% reduction in DNA usage can be acheived. This approach does not require abandoning well-understood transient platforms; it enhances them, delivering immediate COGs relief and throughput gains while preserving flexibility for diverse products and timelines.
Looking forward, successful CDMOs will employ high-intensity, closed, automated and increasingly continuous production systems. That means high-density cell banks enabling condensed seed trains, N-1 perfusion to drive robust inocula, perfusion production bioreactors for higher volumetric productivity and connected downstream purification to reduce hold times and human intervention. Together, these elements compress cycle times, reduce variability and improve line utilization—practical levers that translate directly into lower cost per dose and faster supply.
Critically, technology innovation must be paired with an integrated value chain. FUJIFILM Life Sciences connects specialty reagents and growth factors, cell culture media development, iPSC platforms and CDMO services across mammalian, microbial, vaccines and cell and gene therapy—all the way from drug substance to drug product and finished goods. For developers, this integration streamlines tech transfer, harmonizes quality systems and creates clear accountability from discovery through commercialization.
Additionally, building on a stable, global network matters. With nine sites across the US, Europe and Japan and more than $8.6 billion invested in recent years, FUJIFILM Biotechnologies operates at the scale and resilience required to support early-phase through commercial programs. That network employs over 4,800 people, supports 21 commercial licenses and spans key modalities: mammalian and microbial biologics, gene therapies and cell therapies. For developers, this means uninterrupted access to capacity, flexible technology paths and continuity across geographies.
In viral vectors specifically, experience and breadth reduce risk. Our teams have supported 50+ INDs and produced 250+ GMP batches across AAV (12+ serotypes spanning AAV2, 5, 6, 8, 9, rh10 and novel variants) as well as adenovirus, lentivirus, HSV and other vectors. With clients in eight countries and scales up to 2,000 L, we help programs navigate the realities of moving from early promise to consistent, regulator-ready supply.
The market signal is clear: approvals continue, but the mix is shifting toward larger indications. To meet that demand, manufacturers must deliver tens of thousands of doses at sustainable costs, without compromising safety or potency. High-intensity transient, maturing producer cell lines and data-driven process design provide a practical roadmap. CDMOs that combine these technologies with global, inspection-tested capacity and a partnership-first mindset will help sponsors move faster, lower risk and expand access.
For developers, the question isn’t whether manufacturing needs to evolve, it’s how quickly you can adopt next-gen approaches without derailing timelines. The answer lies in choosing partners who can execute both today’s proven methods and tomorrow’s platforms, validate them under GMP and scale them globally. That’s how gene therapy moves from rare breakthroughs to broad patient impact.
Article top image credit: Permission granted by FUJIFILM Biotechnologies
Gene therapy makers rally after news of Prasad’s exit
As the head CBER, Prasad contributed to an uncertain regulatory climate that depressed gene therapy research. Many developers rebounded on the chance of a more industry-friendly successor.
By: Jacob Bell• Published March 9, 2026
Shares of several gene therapy developers spiked in response to the planned departure of a top agency official who has opposed the ways in which genetic medicines are reviewed.
The Food and Drug Administration disclosed in March that Vinay Prasad, who last May began leading the division that reviews vaccines as well as cell and gene therapies, would be leaving the agency. It’s his second departure in less than a year, as Prasad had stepped back for a few weeks last summer after a major dispute between the FDA and Sarepta Therapeutics that angered high-profile conservatives.
Prasad was a staunch critic of his predecessor Peter Marks, who received both praise and pushback for championing regulatory flexibility as he oversaw the review of COVID-19 vaccines and dozens of cell and gene therapies. During his short tenure, Prasad established stricter approval guidelines for COVID shots. And though he helped outline a new regulatory pathway designed to accelerate the development of treatments for ultra-rare conditions, Prasad has also been directly or tangentially embroiled in a series of surprise delays, rejections and guidance shifts that left drugmakers and industry experts upset and confused about the FDA’s standards.
His planned exit is therefore “a big win for biotech, especially for companies in the rare disease space,” according to Paul Matteis, an analyst at the investment firm Stifel. Genetic medicine developers, which often set their sights on rare diseases, exemplified this, as shares of around two dozen were up immediately after the announcement.
Among the top performers was UniQure, whose stock price had been hammered during a prolonged back and forth with the FDA.
Last September, the company unveiled positive results from a mid-stage clinical trial of a Huntington’s disease gene therapy that impressed researchers and investors, and that UniQure believed were strong enough to earn approval. Executives were therefore taken aback in the fall when, after consulting with the FDA for months, agency staff indicated the data likely weren’t sufficient to support a marketing application.
From there, the outlook for UniQure’s therapy, codenamed AMT-130, only got grimmer. During a late February appearance on CNBC, FDA Commissioner Martin Makary defended his agency’s approach to rare disease drug approvals and made comments that some investors interpreted as him specifically disparaging AMT-130. Shortly after, UniQure disclosed that FDA staff had “strongly recommended” it run another study of its therapy before filing for approval — a development Joseph Schwartz, an analyst at Leerink Partners, called a “worst case scenario for many investors.”
More recently, the FDA took the highly unorthodox step of holding a call between journalists and a high-ranking agency official who spoke about UniQure’s case on the condition of anonymity. Subsequent reports detailed how the official panned AMT-130 and accused UniQure of mischaracterizing its talks with the agency. Many speculated Prasad was the anonymous source. Congressman Jake Auchincloss, D-Mass., posted on social media that Prasad had, in discussing the case, violated agency rules and federal law.
Makary confirmed just hours later that Prasad would be leaving the agency in April, as he’d finished the work he set out to accomplish.
UniQure’s therapy “has evolved into a symbol of FDA inflexibility, or at the very least FDA inconsistency,” Matteis wrote in a note to clients Monday. “This is not to say that AMT-130 approval .... will be a slam dunk. But it does seem like after the dust settles here, it has become much more likely that [UniQure] will get the product review that they were seemingly promised.”
Analysts appear to agree that the big question facing biotech is who will replace Prasad. “We see this news as a double-edged sword,” wrote Brian Abrahams, of RBC Capital Markets, referring to Prasad’s upcoming exit.
While it could have some “direct positive implications” for developers of biologics or gene therapies, it also “perpetuates the regulatory leadership volatility that has kept companies uncertain about their developmental direction and many investors on the sidelines,” Abrahams wrote. Additionally, the replacement, even if more lenient, “would likely still mirror the overarching views the Agency and Commissioner Makary currently have.”
At Stifel, Matteis and his team anticipate that, with midterms later this year, the Trump administration won't want to pick someone controversial, especially with regard to vaccines.
Josh Schimmer, an analyst at Cantor Fitzgerald, hopes the new head of the FDA’s biologics review division will be somewhere between “dovish” or “hawkish.”
“There are costs to an overly-permissive FDA environment just as there are costs to an overly-restrictive one,” he wrote.
Article top image credit: Courtesy of UniQure
UniQure to file gene therapy for approval in reversal reflecting major shifts at FDA
The decision marked another regulatory U-turn following the exits of Marty Makary and Vinay Prasad, suggesting to some analysts that current FDA leadership may be more flexible in certain cases.
By: Jacob Bell• Published June 17, 2026
UniQure, the Netherlands-based biotechnology company, intends to formally ask the Food and Drug Administration to approve its for Huntington’s disease gene therapy now that the two parties are more aligned on the closely watched treatment.
UniQure said in June that, during a recent meeting, FDA staff agreed three years of data gathered from a key trial of the therapy would be enough to support an approval application. As such, the company expects to file one sometime between July and the end of September. The FDA has requested another trial be conducted to confirm the treatment’s effects and, according to the UniQure, the agency wants to make sure both sides see eye to eye on this study’s design before a marketing application gets submitted.
The update is a major about-face for the FDA and a reflection of recent changes in its leadership. Marty Makary resigned as commissioner in mid-May, just days before the firing of Tracy Beth Høeg, who served as acting director of the agency’s main drug review division. Those shake-ups came a little over two months after Vinay Prasad, who oversaw the office that regulates vaccines and gene therapies, departed the FDA for a second time.
Prasad played a particularly important role in what would became a winding, highly publicized saga for UniQure’s “AMT-130” therapy. An oncologist by training, Prasad had been a staunch critic of the ways in which the FDA had tried to expedite the review of certain genetic medicines. Some speculated that, as UniQure received unexpected pushback from the agency, Prasad was either directly or tangentially involved.
UniQure first disclosed plans to file for approval last fall, based on new findings from that key trial. There, researchers evaluated two different dose levels of AMT-130 in 29 participants and compared their disease trajectory to an external control group of similar people in a large, observational study. Results showed that, among 12 patients given the higher dose, signs of disease progression had slowed by 75% after three years.
The data thrilled the Huntington’s community. Investors were equally impressed, as UniQure’s share price more than tripled on the news. The company’s chief medical officer said the findings “reinforce our conviction that AMT-130 has the potential to fundamentally transform the treatment landscape for Huntington’s.”
UniQure leadership was therefore surprised when FDA staff, in a meeting not long after, didn’t view those results as adequate enough to support approval. UniQure noted how that was a “key shift from prior communications.”
From there, the outlook for AMT-130 continued to dim. In late February, during an appearance on CNBC, Makary sought to defend his agency’s approach to approving rare disease therapies. But some investors interpreted certain comments he made as critical of AMT-130 specifically, leading UniQure shares to fall more than 30%.
The following week, UniQure affirmed that FDA staff still had issues with the evidence gathered in its study — the use of an external control group, for example — and “strongly recommended” the company conduct another trial with a control group that receives a “sham surgery.” One Wall Street analyst described this update as the “worst case scenario for many investors.”
The controversy hit a head a few days later, when the FDA took the highly unorthodox step of holding a call between journalists and a high-ranking agency official who spoke about UniQure’s case on the condition of anonymity.
Subsequent reports detailed how the official panned AMT-130 and accused UniQure of mischaracterizing its talks with the agency. Many speculated Prasad was the anonymous source. Congressman Jake Auchincloss, D-Mass., posted on social media that Prasad had, in discussing the case, violated agency rules and federal law. Hours later, Makary confirmed Prasad would be leaving the agency in April, as he’d finished the work he set out to accomplish.
To analysts, UniQure’s announcement Wednesday was a positive sign not only for AMT-130, but other developers who hit unexpected roadblocks from the FDA.
“We think this is great news for [UniQure] and may suggest that the pendulum between regulatory leniency vs inflexible scientific rigor is swinging back to the former now that Vinay Prasad and Marty Makary have left the FDA,” wrote analysts at RBC Capital Markets in a note to clients.
In their own note, William Blair analysts wrote that the change of heart toward UniQure and another drugmaker, Replimune, suggests the current FDA, “largely in caretaker mode, appears to be more flexible on regulatory paths for applications where concerns were previously raised.”
Article top image credit: Sarah Silbiger via Getty Images
Aurora sets out to capitalize on FDA’s new framework for bespoke drug therapies
Led by biotech veteran Ed Kaye, the company intends to simultaneously develop many gene editing treatments for rare conditions by using the agency’s “plausible mechanism” pathway.
By: Ben Fidler• Published Jan. 9, 2026
A biotechnology firm hatched by two prominent researchers publicly debuted in January, aiming to use a new regulatory framework to quickly develop many gene editing treatments for rare diseases.
Called Aurora Therapeutics, the startup was co-founded by Nobel laureate Jennifer Doudna and genetic medicine expert Fyodor Urnov and seeded by Menlo Ventures. It intends to simultaneously work on multiple therapies for the same condition, each of which target different genetic mutations, and quickly advance them with the help of the Food and Drug Administration’s recently unveiled “plausible mechanism” pathway.
Aurora is led by Ed Kaye, a veteran biotech executive and former leader of Sarepta Therapeutics and Stoke Therapeutics. Menlo provided $16 million in seed funding to the startup, which will first test its approach on phenylketonuria, a genetic disease that disrupts the body’s ability to metabolize an important amino acid.
Aurora’s emergence represents the latest attempt to solve a business problem that’s slowing the progress of genetic medicine.
Advances in sequencing technologies and powerful tools like CRISPR have made it possible to rapidly pinpoint and correct genetic errors known to drive thousands of rare diseases. But the small number of patients with those conditions and the exorbitant costs of developing gene-based treatments for them make for a daunting investment proposition, leaving many would-be therapies out of reach.
Notable headway has been made in recent years, though. Academic researchers have, in multiple cases, designed custom therapies for specific individuals. The most prominent example came last year, when researchers at the Children’s Hospital of Philadelphia and elsewhere designed and developed within months a CRISPR-based treatment for a critically ill baby named KJ Muldoon. That treatment has helped stabilize KJ’s condition.
KJ’s story caught the eye of Food and Drug Administration leaders. Last November, former Commissioner Marty Makary and top deputy Vinay Prasad featured it while explaining the plausible mechanism pathway, a regulatory tool meant to speed along treatments for rare and serious conditions. Crucially for gene editing companies, they described how developers could use data from an initial approval — say, for a person whose disease is tied to a specific genetic mutation — to make minor modifications that could support additional clearances for people with other mutations.
Aurora will test that blueprint. Leaning on technical advances enabling the faster design and production of gene editing components, the company is working on what it claimed in a statement would be the first platform to treat the types of mutations historically “impossible to address at scale.”
According to Kaye, Aurora will start out by focusing on a disease’s more frequent mutations. Doing so would enable it to get a “commercial foothold that generates revenue” while building a way to efficiently develop subsequent therapies for rarer mutations.
“Eventually, we could get to a point where we could very quickly respond to individual patient needs,” as was the case with baby KJ, Kaye said. “But we first really need to have a well-developed system that is sustainable before we go after very, very rare indications.”
Aurora believes it can build that system by starting with phenylketonuria, or PKU. The disease is caused by a wide range of mutations to a gene called PAH. While it’s currently managed with lifelong therapy, dietary restrictions and monitoring, those approaches aren’t curative and can still leave patients with cognitive deficiencies.
PKU is also well-understood biologically and, as a result, long been a target of genetic medicine. But unlike previous efforts, Aurora is designing multiple therapies. The first will simultaneously address the three most common PKU-causing mutations. Aurora will then broaden its approach and add more.
Kaye believes that, as a result, the disease “fits in the plausible mechanism pathway perfectly.”
“We understand the proximate cause of the disease, and we know how to address it,” he said.
Article top image credit: Permission granted by Aurora Therapeutics
FDA approves Regeneron’s hearing loss gene therapy
Otarmeni was the first gene therapy cleared under the FDA’s “national priority” voucher program and is being offered to eligible patients at no cost.
By: Jacob Bell• Published April 23, 2026
For the first time, people born with a rare, inherited form of deafness have a treatment option beyond hearing aids and cochlear implants.
That’s because, in April, the Food and Drug Administration approved a one-off therapy designed to overcome the genetic hiccup that causes this hearing loss and offer a lasting fix. Developed by Regeneron Pharmaceuticals, the gene therapy has already shown great promise in the key clinical study supporting its approval.
“I’ve witnessed firsthand my trial participant responding to their mother’s voice, dancing to music and interacting with the world, and these moments are now possible for more children born with this specific form of hearing loss,” said Eliot Shearer, an investigator in that trial, in a statement from Regeneron.
The therapy, Otarmeni, was cleared for use in children and adults who have severe-to-profound hearing loss because of variations in a gene known as OTOF. Such variations keep hairs in the inner ear from producing a protein that’s essential for relaying sound signals to the brain. Otarmeni is administered through a surgical process similar to that of cochlear implants, and uses a disarmed virus to smuggle into the inner ear a working copy of the defunct gene.
That key study recruited 20 children with OTOF hearing loss, who ranged in age from 10 months to 16 years and were given a dose of Otarmeni in either one or both ears. Four participants entered already having a cochlear implant in one ear, while some others got the devices as part of the experiment. The main goal was to see if Otarmeni could get hearing into the moderate to normal range.
Six months after treatment, 80% of participants had hit that benchmark. According to Regeneron, they could hear at least 70 decibels — the kind of sound one might expect from a loud conversation or a clamorous household appliance. Whether a patient was younger or older also didn’t seem to impact the therapy’s effectiveness, a result that likely influenced the FDA to include adults in the label.
“Is there really any difference between a 16- and a 21-year-old for something like this? We don't think so,” said Jonathan Whitton, Regeneron’s vice president of genetic medicines, in an interview. “It's a pleasant surprise for a lot of people in our field, me included, that it looks like the treatment window is so broad.”
In the trial, parents of the 10-month-old noticed changes within a few weeks. Their daughter started reacting to handclaps and became more aware of speech and whispers. Parents of a different young girl reported “spectacular” progress one year after treatment, as she could now respond to distant sounds and understand speech in noisy environments.
So far, Otarmeni also appears generally safe, with the most common adverse events being inner ear infections, vomiting, nausea, dizziness and procedural pain. Lawrence Lustig, another trial investigator, noted after a data release late last year how these events were mostly minor, short-lived and the type to be expected from any sort of ear-related surgery.
Otarmeni has “rapid, meaningful and consistent” effects, and its approval “signals a new era in the treatment of genetic forms of hearing loss, where reinstating 24/7 natural hearing is now possible,” added Shearer, who also serves as an otolaryngologist at Boston Children’s Hospital.
As with most cutting-edge medicines, there are still outstanding questions. Scientists aren’t entirely sure how long Otarmeni’s effects will last, or why it works better in some patients. At that earlier data cut, at least one participant didn’t have any significant hearing improvement. There, Whitton said none of the most likely causes have checked out. The leading hypothesis now, he said, is there was some kind of delivery issue, but “we don’t have definitive proof of that yet.”
Regeneron said it will provide Otarmeni at no cost to “clinically eligible individuals” in the U.S., though that doesn’t necessarily mean no out-of-pocket costs for administration. In the past, insurance companies have seriously weighed the benefit and durability of gene therapies, which often carry list prices into the hundreds of thousands — if not millions — of dollars.
“What we’re talking about, really, is: what’s the value of hearing?” Whitton said in a previous interview. “My expectation is most people see massive value, having hearing connect you to your environment, to others, to your families.”
Yet that value isn’t guaranteed to translate to commercial success. Very few people have OTOF-related hearing loss. The addressable population is further winnowed by the fact that Otarmeni can’t be administered to any ears that already have cochlear implants.
David Risinger, an analyst at the investment firm Leerink Partners, has also described a “crowded competitive landscape” in this area of drug development, with at least three companies, including Eli Lilly, advancing their own gene therapies.
Otarmeni received a lightning fast approval review thanks to a new and controversial voucher program recently enacted by the FDA. It's the first gene therapy with a “national priority” voucher to be OK'd for market.
Article top image credit: Getty Images
Intellia CRISPR drug succeeds in late-stage study against rare swelling disorder
The findings position Intellia to bring to market the first “in vivo” gene editing medicine, though the therapy’s commercial potential remains the source of intense investor debate.
By: Ben Fidler• Published April 27, 2026
An experimental gene editing medicine from Intellia Therapeutics succeeded in a Phase 3 trial, positioning the company to seek approval of what would be the first treatment of its kind for a rare disorder known as hereditary angioedema.
When compared to a placebo, the therapy, “lonvo-z,” reduced the rate of the disease’s hallmark swelling attacks by 87% over the course of about six months, meeting the study’s primary objective. Lonvo-z also helped rid 62% of recipients of disease attacks or the need for other therapies during that follow-up period, versus 11% of placebo patients.
Intellia said, without specifics, that lonvo-z had a “favorable” safety and tolerability profile. The most common treatment-emergent side effects were infusion-related reactions, headache and fatigue, and all reported by a Feb. 10 data cutoff were mild to moderate in degree. The company has begun a “rolling” U.S. approval submission and, assuming a clearance, intends to launch lonvo-z in the first half of 2027.
Intellia was one of the first biotech companies built around the Nobel prize-winning, gene editing technology known as CRISPR.
Lonvo-z is a microcosm of that journey. Intellia billed the therapy as a possible “functional cure” for hereditary angioedema, or HAE, a rare condition that causes swelling attacks that can affect multiple organs. As a one-time, long-lasting treatment, lonvo-z might free people from relying on frequent injections or pills to prevent or treat symptoms.
But genetic medicines have struggled to sell in areas where, as with HAE, multiple effective medicines already exist. Therapies like lonvo-z also carry the specter of certain safety problems — like accidental, off-target edits — that more traditional drugmaking technologies don’t. Those factors have led some analysts to suggest Intellia’s therapy may need to prove exceptionally potent, durable and safe to entice patients and physicians to try it.
The data revealed in April give Intellia the chance to make that case. Study results showed that, during the treatment period, the average monthly attack rate among lonvo-z recipients was 0.26. That equates to roughly three attacks a year, compared to just over 2 a month for those on a placebo.
Moreover, all patients given lonvo-z in the study were still off other preventive therapy as of that mid-February data cutoff.
Importantly, Intellia didn’t observe the kind of liver-related side effects that have slowed another treatment it’s developing for a rare heart condition. This “clean safety” addresses a key concern among investors on the program and company “as a whole,” wrote Leerink Partners analyst Mani Foroohar. The therapy’s efficacy is “competitive” with CSL’s preventive drug Andembry and could “strengthen over time” with longer follow-up, Foroohar added.
Yet the skepticism about lonvo-z’s sales potential persists. The Food and Drug Administration in the last year has approved multiple newtherapies for HAE, giving patients several ways to keep their symptoms at bay.
The ongoing “debate” is now shifting from clinical data to lonvo-z’s “place in the treatment paradigm,” Foroohar wrote.
Evercore ISI analyst Jonathan Miller previously projected that peak annual sales will top out at $500 million, given the way gene therapy launches have gone so far.
Article top image credit: Permission granted by Intellia Therapeutics
Sarepta, the FDA and a Duchenne gene therapy crisis
Sarepta eventually consented to the FDA’s request to stop selling Elevidys, but the company’s brief standoff with the agency could still carry major consequences for the Duchenne community.
By: Ben Fidler, Ned Pagliarulo• Published July 21, 2025• Updated July 22, 2025
Editor’s note: Sarepta Therapeutics said July 21 it is temporarily halting shipments of Elevidys, reversing its prior refusal of a Food and Drug Administration request. Read our story for updated information.
In June, a 51-year-old man treated in a clinical trial with an experimental gene therapy became dangerously sick. The developer of that treatment, Sarepta Therapeutics, informed the Food and Drug Administration his case could be life-threatening.
The man died from acute liver failure a few weeks later, which Sarepta reported to the FDA on July 3 as a matter of course. Little has seemed to go by the book since.
Liver injury is a known risk of the kind of gene therapy used to treat the man, who had a muscle-weakening disease called limb-girdle muscular dystrophy. Two other patients with a different kind of muscular dystrophy, Duchenne, and treated with a different Sarepta gene therapy, approved as Elevidys, also died of liver failure this year. In response to those earlier deaths, Sarepta stopped shipping Elevidys to certain older Duchenne patients.
But Sarepta didn’t consider the 51-year-old man’s death to be “material,” a regulatory term describing when an event is important enough to require public disclosure. The death went unreported publicly until July 17, one day after Sarepta had held a conference call to discuss a business restructuring that will shelve the limb-girdle treatment, along with several others.
Wall Street analysts who cover Sarepta were furious over the lack of transparency, forcing Sarepta to hastily hold another conference call, on July 18, to explain why it hadn’t disclosed the man’s death, which raises serious questions about the safety of that gene therapy and, potentially, Elevidys.
The FDA appeared angered as well. Despite knowing about the most recent patient death for weeks, and having already discussed Elevidys labeling changes with Sarepta, the agency on July 18 asked Sarepta to stop shipping the drug to Duchenne patients. In statements, the FDA implied it was looking at a common component between Elevidys and the experimental therapy.
Sarepta initially refused the FDA’s request, saying it will continue shipping Elevidys for younger Duchenne patients who can still walk — a group for whom the company claims treatment remains supported by available evidence. Three days later, it begrudgingly agreed.
The standoff between Sarepta and the FDA, while brief, has few precedents. Analysts see rising risk the FDA formally attempts to withdraw Elevidys from market, which could divide a patient community that has for years helped Sarepta push for greater regulatory flexibility at the agency. For the gene therapy field, meanwhile, the brewing crisis comes at a time when investment has dried up and its future appears fragile. Here are five questions about what might come next:
What does Elevidys’ future look like now?
Some backstory first: The FDA first approved Elevidys in June 2023, granting it accelerated clearance for Duchenne patients who were 4 or 5 years old and could still walk. One year later, the agency expanded the treatment’s OK to include ambulatory and non-ambulatory Duchenne patients who were 4 years of age or older.
Both decisions were controversial, as Sarepta’s trial data for Elevidys didn’t prove the therapy could significantly improve motor function. But the Duchenne community mostly embraced Elevidys, propelling it to the fastest market launch of any gene therapy approved in the U.S.
Safety concerns prompted by the recent deaths could now change that. But without Sarepta’s cooperation, the FDA’s options for stopping Elevidys sales altogether are limited. And those it does have at its disposal are likely to take time.
In first refusing the FDA’s request to halt sales, Sarepta maintained there has been no new information indicating greater safety risk in younger, ambulatory patients. Both of the Elevidys patients who died were teenagers whose disease had eroded their ability to walk.
The distinction is clinically important as Elevidys is dosed by weight, so younger, lighter patients receive less of the drug. And as they’re earlier in their disease course, the treatment window to forestall further damage may be greater.
The distinction also has regulatory implications. In June 2024, when the FDA expanded Elevidys’ approval it did so by converting its accelerated approval to full for ambulatory patients. In non-ambulatory patients, Elevidys’ clearance remains “accelerated,” which indicates Sarepta is required to produce further evidence of its benefit in this group.
The FDA has more tools at its disposal to request market withdrawal of therapies under accelerated approval than it does for those under traditional OKs. But even here, its power to compel drugmakers to stop sales of a product is attenuated by a process requiring regulatory notice, comment periods and public hearings.
Analysts seem to think the FDA might still try. “We don't believe FDA is likely to let go without a fight — and while we can't predict timing (not much precedence in this occurring), there is a good chance the FDA at some point forces this from the market,” Cantor Fitzgerald analyst Kristen Kluska wrote in a recent client note. — Ned Pagliarulo
Why did the FDA wait to step in?
The FDA claimed on July 18 that it took “swift action” by suspending multiple Sarepta trials and requesting the company halt all Elevidys shipments. It took those steps following “new safety concerns” that the agency said showed patients may be exposed to “unreasonable and significant risk of illness or injury.”
“We believe in access to drugs for unmet medical needs but are not afraid to take immediate action when a serious safety signal emerges,” FDA Commissioner Martin Makary said in the statement.
According to Sarepta, the FDA had known about the latest death, which occurred in June, for two weeks. But the FDA didn’t take action until July 18, after news of the death became public through reports and a conference call from Sarepta.
There were “no new or changed safety signals” regarding Elevidys, Sarepta added, noting how the most recent death occurred with an experimental therapy that’s manufactured differently and administered at a different dose. Both treatments use the same viral vector for delivery into the body, however.
Sarepta was already under fire for waiting weeks to disclose the most recent death, a decision CEO Doug Ingram last week said was because the company believed it was “neither material nor relevant.” Its share price has been decimated as a result.
Yet the agency’s delayed response was “puzzling” and “perplexing,” according to analysts on Wall Street. They suggest the FDA reacted only in response to “recent headlines,” wrote Leerink Partners’ Joseph Schwartz, and hint at a “shift towards decision-making influenced more by social factors than by science or regulatory precedent.”
“This makes it increasingly more difficult to anticipate how [the FDA] will govern moving forward,” Schwartz added, noting how the “optics” look bad “both for the agency and the company.” — Ben Fidler
Where does Sarepta go from here?
Sarepta posted net yearly losses for much of its history, but finally became profitable in 2024 because of Elevidys. The company expected that financial success to continue in the coming years, predicting in February that its overall revenue would reach $2.9 billion to $3.1 billion in 2024. Analysts anticipated Elevidys would surpass $2 billion on its own, enabling Sarepta to afford pursuing more than 40 programs based on gene therapy, gene editing and RNA technologies.
Those projections have quickly fallen apart. Sarepta revised and suspended its financial guidance after halting Elevidys shipments to non-ambulatory patients and, last week, cut nearly 40% of its workforce along with several drug programs. Gene therapy research, once a main focus, has been whittled down to a single prospect for limb-girdle muscular dystrophy.
The biotechnology company is now counting on a trio of “exon skippers” for Duchenne, which it suggested could bring in $900 million annually, and Elevidys, which it said at minimum should generate about $500 million per year. An RNA-focused collaboration with Arrowhead Pharmaceuticals could yield more medicines in the future.
But Sarepta could face more financial peril. It has about $1 billion in debt due in 2027 and intends to speak with lenders this week to determine whether any of the myriad recent developments might constitute a “material adverse event” that could trigger financial fallout, executives told Leerink’s Schwartz. The company would need to make “sizable organizational cuts” to pay its debts and remain viable if Elevidys were to be removed from the market entirely, Schwartz wrote.
Even now, investors are wondering whether Sarepta can pay its debts without more cuts, Schwartz added.
The company could face competitive threats to its exon-skipping drugs as well as Elevidys in the near future, further threatening its core business. And the clinical holds announced by the FDA will at minimum likely delay the arrival of its next-closest product to market, a gene therapy for limb-girdle dystrophy, wrote William Blair analyst Sami Corwin on Monday. — Ben Fidler
How will the Duchenne community respond?
Several Duchenne medicines have been approved by the FDA over the last decade or so.
These medicines have offered hope to a community of patients, caregivers and advocates facing a lethal disease that slowly robs children of their ability to walk and function independently. They’ve also tested the FDA, spotlighting the tricky decisionmaking involved in evaluating treatments for rare, deadly conditions and the pressure patient groups — many of which receive funding from drug companies — can put on regulators.
That community is now at the center of a high-stakes standoff between a company and its regulator. In the last few months, they’ve heard of multiple deaths in clinical trials, potentially changing how they see the risk-benefit balance of Elevidys. They’ve seen access for Elevidys restricted and clinical trials put on hold. And now, they’ve received conflicting reports from Sarepta and the FDA about Elevidys’ market status.
“Families who have fought tirelessly for access to this therapy, those who have already received it, and those who are in line to receive it are now left with more questions than answers,” wrote Parent Project Muscular Dystrophy, a prominent patient advocacy group, in a July statement. PPMD is “urgently seeking answers and calling on both Sarepta and the FDA to provide clarity and transparency.”
“These developments are painful, and they’ve shaken the hope that many families have carried for years,” added the Muscular Dystrophy Association, another large advocacy organization, in a separate statement in late July.
Should the FDA try to withdraw Elevidys altogether, it could start a messy fight with an educated and mobilized patient community that has influenced past agency decisions — the kind of spat an optics-conscious agency might want to avoid.
Such a move would also counter some of the messaging from Makary and Vinay Prasad, who now runs the FDA’s Centers for Biologics Evaluation and Research. Prasad was highly critical of his predecessor Peter Marks’ decision to twice overturn agency staffers in approving and then broadening use of Elevidys. But, like Marks, he and Makary have promised FDA flexibility in reviewing rare disease gene therapies. — Ben Fidler
What are the implications for the gene therapy field?
The patient deaths, and Sarepta’s handling of them, come as the gene therapy field faces its own crisis.
Developers of these medicines are having trouble demonstrating they can reliably profit off of their sale. Logistical, financial and administrative hurdles are hampering adoption of approved therapies, which in turn is crimping the outlook of those in development. The slump in investment that’s holding back biotech writ large is hurting gene and cell therapy companies even more acutely.
This newest episode presents two challenges. Firstly, the patient deaths bring the surface safety concerns that have stayed simmering underneath gene therapy ever since the 1999 death of Jesse Gelsinger in one of the field’s early clinical trials. Elevidys, and Sarepta’s experimental limb-girdle treatment, use a type of engineered virus — called AAV for short — that’s become a go-to method for delivering functional genes to target cells. Researchers have warned, though, that high doses of these therapies present notable risks, which now appear back in the spotlight.
Cantor Fitzgerald analyst Eric Schmidt wrote in a July 18 note that he’s heard concerns from investors and companies that Sarepta’s decision to discontinue swathes of its gene therapy research “would be another difficult setback for the AAV platform.”
Sarepta’s lack of disclosure may carry consequences of its own, too. “Fears are running high that Sarepta’s downfall could be a Theranos/Vioxx/Gelsinger-type of event that further poisons generalists against the sector,” Schmidt added, referencing how biotech safety scandals of the past led to declines in investment.
“In what other industry can a company's revenue projections drop from ~$5B to near nothing in weeks?” he added. “Where else are investors so reliant on management to provide accurate information? And how can an industry that claims to put patients first have gone so far afield?”
Sarepta’s credibility problems are of its own making, of course, but for a sector that desperately needs investors to regain confidence in its potential, there may be knock-on effects. — Ned Pagliarulo
Article top image credit: Courtesy of Sarepta
BioMarin, following sluggish sales, withdraws hemophilia gene therapy
Roctavian was pulled from the market following slow uptake and a failed search for a buyer.
By: Ben Fidler• Published Oct. 27, 2025
BioMarin Pharmaceutical is giving up on the hemophilia gene therapy Roctavian, announcing in February plans to withdraw a first-of-its-kind medicine once expected to become a future blockbuster.
In an earnings report, BioMarin said a “comprehensive effort” that began in October failed to unearth a “qualified buyer” for Roctavian, leading the company to pull it altogether.
The announcement culminates what’s been a fast fall for Roctavian since its launch began four years ago.
Roctavian’s approval in Europe in 2022 and in the U.S. a year later were scientific milestones, the culmination of years of research developing a genetic medicine for hemophilia A. As a one-time, long-lasting treatment, Roctavian was billed as an alternative to the chronic therapies people with hemophilia A use to prevent bleeding. It was also seen as a clear example of the potential economic bargain of a gene therapy that, despite a high initial price tag, might alleviate the need for supportive care patients would receive instead.
At the time, many Wall Street analysts viewed the product as a blockbuster-to-be. Leerink Partners analysts once projected $2.2 billion in peak sales, and BioMarin was similarly optimistic, estimating early on that the therapy would generate anywhere from $50 million to $150 million in 2023.
Instead, Roctavian has become a cautionary tale of the challenges drugmakers can face selling a gene therapy. BioMarin quickly and sharply slashed its revenue forecasts for 2023 and ended up recording $3.5 million in product sales that year. The therapy accounted for only $26 million in 2024, and just $23 million over the first nine months of 2025, the company said Monday.
CEO Alexander Hardy has previously cited the “complexity” of getting patients on treatment as a reason for Roctavian’s commercial performance. But doubts about the durability of its benefits and a price tag that made reimbursement discussions challenging also slowed its sales trajectory.
In August 2024, BioMarin pared down Roctavian spending rather than choosing to sell it altogether, with Hardy at the time citing signs of launch progress in the three countries — the U.S., Germany and Italy — the company chose to focus on. Those plans changed in October, when the company revealed plans to “pursue options to divest Roctavian and remove it from our portfolio.”
Article top image credit: Getty Images
‘No tolerance for failure’: An oral history of the first CRISPR medicine
A new sickle cell disease therapy developed by CRISPR Therapeutics and Vertex Pharmaceuticals is now approved in the U.S. and U.K. This is the story of how it came to be.
By: Ned Pagliarulo• Published Dec. 10, 2023
The last line of this century’s most important biomedical research paper contained a hint of the scientific revolution to come. An ancient bacterial defense system, the researchers wrote in 2012, could be adapted to offer “considerable potential for gene targeting and genome editing applications.”
The past decade has proven those words a dramatic understatement. The bacterial defense system, dubbed CRISPR, is the foundation for a flexible and powerful gene editing tool that’s allowing scientists to reimagine how to treat disease. A new generation of biotechnology companies has come of age translating that research into medicines that can turn genes off or on, or even rewrite DNA code directly.
“I think CRISPR is one of the most fundamental innovations in life sciences we have seen over the last 20 years,” said Rodger Novak, co-founder and former CEO of CRISPR Therapeutics, one of the first biotechs formed to develop CRISPR-based drugs.
For people with sickle cell, the future is now. On Nov. 16 and Dec. 8, regulators in the U.K. and U.S. approved Casgevy, a near-curative treatment developed by CRISPR Therapeutics and Vertex Pharmaceuticals for the inherited blood condition. It’s the first CRISPR gene editing medicine to win clearance for commercial use.
Casgevy’s journey to approval is a remarkable story of scientific discovery, bold bets and steady perseverance. To Stuart Orkin, a professor of pediatrics at Harvard Medical School whose research outlined how CRISPR could be used to treat sickle cell, it is a “great example of the way things should go.”
“In academia, we do discovery. The role of pharma and biotech, in my view, is to take these discoveries and bring them to patients,” said Orkin. “We made our discoveries. They did the trials. They didn’t mess it up.”
CRISPR Therapeutics and Vertex’s achievement with Casgevy happened more quickly than is usual in biotech, where scientific breakthroughs are often only the beginning of a long and arduous process. Alnylam Pharmaceuticals, the pioneer of a gene silencing method of drugmaking known as RNA interference, needed 16 years to turn an academic discovery into the first RNAi medicine. Casgevy’s first approval, by comparison, came 10 years after CRISPR Therapeutics’ founding.
“It happened a lot faster for gene editing,” said John Maraganore, Alnylam’s founding and now former CEO. "Opening up the door for a new modality is an epic moment."
Casgevy’s success wasn’t a sure thing, though. The drug’s story is also one of patent battles, safety scares and stock gyrations. Other companies tried to apply CRISPR to sickle cell, but came up short.
This oral history of Casgevy’s development is based on nearly two dozen interviews with the scientists, executives, physicians and sickle cell patients who helped make the medicine a reality. All titles are presented based on the principal relevant roles held by speakers during the time of each chapter, unless unchanged. Interviews have been condensed and edited for clarity.
CRISPR researcher Emmanuelle Charpentier stands in a laboratory at the Helmholtz Centre for Infection Research in Braunschweig, Germany, on May 19, 2015.
Peter Steffen/Picture-Alliance/DPA/AP
Chapter 1 The Beginnings (2012 - 2015)
The discovery of CRISPR by Emmanuelle Charpentier, Jennifer Doudna, Feng Zhang and others sparked a frenetic race to capitalize on the technology’s potential. Scientists, investors and executives set about to construct business plans for building new gene editing companies — work that resulted in the creation of Caribou Biosciences, CRISPR Therapeutics, Editas Medicine and Intellia Therapeutics.
While it was clear to many that CRISPR was an important new tool, there was disagreement over how quickly and broadly it could be applied, or even how it compared to existing editing techniques like zinc fingers and TALENs.
Simeon George (CEO, SR One): It was 2012 when the first paper from Charpentier and Doudna was published. Within the first six, 12 months, there was clearly a sense that this could be transformative. It had the potential to have this laser-guided approach to treat, repair and possibly cure. Even from those early days, it looked like a step change from everything we'd seen before. There was immediately this sense of wonder around the technology.
Rodger Novak (co-founder and CEO, CRISPR Therapeutics): But it was certainly not the case that the industry, in particular VCs, were all over CRISPR when the paper hit in 2012. There were some believers. But it was so early, it was actually pretty difficult in the beginning to convince people that this is real.
Rachel Haurwitz (CEO, Caribou Biosciences): Both zinc fingers and TALENs had left investors convinced that genome editing is a really hard thing. At that point in time, you basically had to have a PhD in genome editing to do it. There was a fundamental skepticism that this was any different.
Even among those who were willing to think a little more creatively, many expected only one or a small number of relevant use cases, not this incredibly broad toolbox. To be quite honest, what has panned out far surpasses the picture I was capable of painting back then. And yet, the picture I painted was far too vast; people didn't think it could be real.
CRISPR Therapeutics co-founder Rodger Novak
Permission granted by CRISPR Therapeutics
Rodger Novak: If we had had CRISPR alone, this would have been tough. But messenger RNA was out there and we had much cheaper gene sequencing opportunities. Technology-wise, around that time, there was light on the horizon and things came together nicely.
Nessan Bermingham (entrepreneur in residence, Atlas Venture): If CRISPR had been there 10, 15 or 20 years before, I'm not sure we would have gotten the attention that we got at the time, because, if you think about genetic medicines, there had already been so much work done. Go back to things like small interfering RNA therapies or antisense oligonucleotides. Go back to what was going on with gene therapy. People were able to connect the dots.
Twenty years before, we didn't have the technologies that were required to allow us to move so rapidly. The timing was very fortunate.
Shaun Foy (co-founder, CRISPR Therapeutics): When we were speaking with scientists and drug developers in the beginning of 2013, all of them got the technology. There were different views on whether it would be translated in a decade or sooner. A lot of people thought it would take much longer than what transpired.
When it came to pitching though, we really didn’t have to pitch for the seed. I reached out to Nessan and he wanted to give me a term sheet right away. And I was speaking with Jerel Davis, and Versant [Ventures] got it fairly quickly.
Nessan Bermingham: Shaun reached out to me and said, ‘What do you think?’ I basically said, ‘We'll give you a term sheet.’ He had been working with Versant and flagged it to them at the same time.
Shaun Foy: It was very complicated at the time: exciting new technology; a number of important experts who were circling around different companies; a number of different investors who were interested in building companies; very complicated sets of personalities and a complicated intellectual property landscape.
We were pretty focused when it came to the investors. I knew we were going to work with Versant and/or Atlas. We never really entertained conversations with any other investors.
Nessan Bermingham: We put a term sheet down and Versant put a term sheet down. Things got to a point where it was clear that Versant and Atlas had slightly different visions on how to build the company moving forward, leading to a more competitive position as to who would lead the deal.
I spent a tremendous amount of time with the team and started to build out the overall initial strategy for the company. One of the areas that they really were focused on was ex vivo applications, which has led to [Casgevy]. But I really felt we should be moving in vivo also.
Shaun Foy: We had thought that Atlas would be part of the seed financing in October 2013 but in the end this didn't happen. Versant were very keen to finance the story initially themselves and, although Atlas had done a lot of work on the story, we actually had much deeper relationships within Versant so this made sense to Rodger and myself.
Atlas continued to work with the story on the basis that they would join in the [next] round. By the time that came around several months later Versant had decided they wanted to take the entire round themselves and in the end we didn't get Atlas into the deal. Nessan went off to found Intellia.
Nessan Bermingham: Ultimately, Versant paid more than we were willing to pay and structured the deal in a way that was attractive to the founders also. And we, Atlas, decided to step out of the process.
Fast forward, literally weeks, we then reached out to Caribou and that ultimately led to the formation of Intellia.
Shaun Foy: We didn't need to pitch anybody else until the end of 2014. By then, we were totally in this CRISPR craze. Every group is knocking on your door. We had something like 10 lead investors that were offering term sheets to cut each other out.
Simeon George: After the first meeting with the founders of CRISPR Therapeutics, it was clear this was the company to invest in. Everyone was using the same technology, by and large. But with these founders the vision was: ‘We want to be the first company to actually develop a medicine.’ Rather than falling in love with the science, they were crystal clear around product development.
From that point on, it was about the financing. How do you cobble together a round? There were questions around IP. There were questions around doing a partnership early on. Ultimately the round that we led, the Series A, frankly looks like it should have been a no brainer. Why would you not invest? But it wasn't an easy round to put together.
Shaun Foy: I really believed at the beginning that [CRISPR] should be one company, and that we should have the key scientists that were involved in the technology, patent estate all under one roof. But over the course of the spring and summer of 2013, it was pretty clear that there was so much interest in the technology that people wanted to do their own thing.
In hindsight, it’s been better for patients to have multiple companies working on this. It’s hard to imagine how one company could get all the work done efficiently.
A scientist at Vertex Pharmaceuticals works in a laboratory in San Diego, California, in 2015.
Gregory Bull/AP
Chapter 2 Charting a course (2014 - 2016)
Thousands of human diseases are caused by genetic mutations. In theory, CRISPR gave researchers and drugmakers the means to fix many. But which to pick? That choice faced CRISPR Therapeutics, Editas and Intellia early on.
CRISPR Therapeutics’ first choices were sickle cell and beta thalassemia, diseases caused by errors in the genetic code for hemoglobin, a vital oxygen-carrying protein. Prior to CRISPR’s discovery, Stuart Orkin had identified a gene, BCL11A, that offered a way to treat both conditions, presenting CRISPR Therapeutics with a valuable roadmap.
Rodger Novak: CRISPR was an entirely new technology. It was not, as some people illustrated, just like editing a Word document. There are so many more complexities. So one thing we asked at the very beginning was, what indication could we go after that reduces complexity while addressing a really important unmet medical need?
Shaun Foy: We wanted to focus ex vivo and on knockout strategies for a variety of reasons, but [mainly] to remove technology risks.
We were trying to identify diseases where there's high unmet medical need and where you could edit a small number of cells and have a big impact. And the two diseases that we looked a lot at were knocking out CCR5 in HIV and knocking out BCL11A. We even had initial conversations with Gilead about the idea of knocking out CCR5.
Rodger Novak: We engaged a firm focused on questions like this. Three or four months later, after a lot of interviews, they came back with a proposal of 10, maybe 20 different indications. I just recently looked back at this and not a single one did we pick. It’s a good lesson learned.
So we came up with sickle cell and beta thalassemia. The next challenge was the board, which said, ‘Oh my God, how can you do that? Look at Editas. Look at Intellia. And then there's Bluebird bio.’ In the end I convinced them that the main focus was simplicity.
You need to differentiate one way or another. It’s relatively irrelevant who's out there otherwise. You should know about it; you should educate yourself. But if you believe in your approach, and you truly believe you can differentiate, then go for it. And that’s what we did.
Bill Lundberg (Chief scientific ofifcer, CRISPR Therapeutics): We didn't start with awe over our brilliant technology platform. We started with: What's the medical problem that we want to try to solve? Let's really truly understand it. Fifty years of sickle cell and beta thalassemia research have really shown what that problem is. It's one of the best understood diseases.
Sam Kulkarni, then CBO of CRISPR Therapeutics, and Bill Lundberg, formerly the company’s chief scientific officer, stand by a poster at the American Society of Hematology’s annual meeting in 2016.
CRISPR Therapeutics
Simeon George: The company and founders were looking very closely at a number of applications. We were also waiting and trying to think about how to prioritize ‘low-hanging fruit.’ Frankly, there isn't low-hanging fruit when you're bringing a new technology forward.
Everything looks obvious in hindsight. At the time, there was risk. We had to take a leap. You don’t know what you don’t know.
Sam Kulkarni (Chief business officer, CRISPR Therapeutics): We’ve seen a lot of platform companies live and die by the choices they make on the initial indications they go after. It wasn’t clear in 2014 and 2015 what was going to work and what wasn’t.
It was clear ex vivo was more likely to work and sickle cell fit because the genetics were known.
Bill Lundberg: We fully understood [sickle cell and beta thalassemia’s] disease biology and pathophysiology. We knew what we had to change. There were all these human genetic variants showing, if their genetics are just a little different, the patients are better. Then it was just an engineering question.
[President John F.] Kennedy challenged the country to get a man on the moon by the end of the decade. He knew he could do that, because he knew the discoveries were solved. All you had to do was just strap five Saturn V rockets together and you’d get there. That’s where we were. We had all the pieces.
Rodger Novak: Without the innovation from academia, [there was] no chance at the time. At the same time, we did build out within CRISPR a very strong research organization. We came to the conclusion that if we don't master the technology and rely almost exclusively on innovation out of academia, we don't stand a chance.
Bill Lundberg: We weren't sure where to edit. And we did these incredibly large experiments looking at a large number of possibilities. We had to simultaneously optimize for a number of different characteristics. Which guide do we use? Do we make synthetic guides? Do we transcribe? Do we need to put some funky nucleotides on the front? How do we introduce it into cells?
We didn’t know. It came down to two different approaches. Ultimately, we chose the one we chose, for various reasons, but we didn’t know that until the very end. We were simultaneously parallel processing everything up until the time we needed to lock down processing and manufacturing. We had huge arguments over whether to spend extra millions of dollars.
On the financing side, for the first year or so, we had a lot of headwinds. Investors were like, ‘This is a Nature paper, why is it going to work? Look, another Science paper, but this isn't a medicine.’ And then it flipped. Suddenly, we had a huge amount of tailwinds. That subsequent financing climate was really helpful.
Tirtha Chakraborty (Head of hematology, CRISPR Therapeutics): Once the platform was built, we [started] the therapeutic work. When preclinical development started, pharmacological models didn't exist. We had to build them from scratch. If I think about how many things didn't exist before this program started, it's quite unreal that it all happened.
Bill Lundberg: [With] a really complicated system, we needed to to simplify [it]. Any complexity equals risk. It really came down to taking as many risks off the table, [such as by] taking cells out of the body.
Simeon George: I felt in those early days that, relative to our small peer group — the other companies that were in Boston, that were well capitalized, that had a lot of noise around them — we were the odd one out. I didn't feel like we got the same level of attention or that there was the same excitement in those first few years. And in part [that was because] we were coming out of Europe.
Bill Lundberg: The pressure came from the other companies who were claiming they were going to radically revolutionize the world. It was clear to me we needed to put our head down. But then it was like, ‘Oh, wait, Editas is going to cure every disease and they're starting with eye diseases. Maybe we should go into eye diseases.’ That kind of pressure is hard. It takes real commitment. You have to trust the reasons why you’re going down the path, stay focused and continue to deliver.
Sam Kulkarni, CEO of CRISPR Therapeutics, speaks in front of employees at the opening of the company’s former office in Cambridge, Massachusetts in 2017.
CRISPR Therapeutics
Chapter 3 CRISPR’s ‘wild ride’ (2015 - 2019)
The promise and acclaim of CRISPR gene editing meant added scrutiny for CRISPR Therapeutics and its peers. The first few years of the biotechs’ existence were dogged by a bitter patent battle between the University of California, Berkeley, and the Broad Institute of MIT and Harvard over who invented the technology. The field was divided in two camps, with CRISPR Therapeutics and Intellia aligned with Berkeley, and Editas with the Broad.
The companies also faced questions about gene editing’s implications for society, and skepticism from investors as they prepared initial stock offerings. Once public, their stocks rose and fell on news of academic findings suggesting safety concerns to CRISPR, or on delays in their efforts to reach clinical trials.
Simeon George: In the early years, the IP estate and how we were prosecuting it was a meaty topic. We all felt it was going to get resolved. If you look to the history of monoclonal antibodies and how this plays out, generally speaking companies can coexist, whether it's by cross licensing or specific composition [patents] that give you coverage for targets.
CRISPR Therapeutics CEO Sam Kulkarni
Permission granted by CRISPR Therapeutics
Sam Kulkarni (CEO, CRISPR Therapeutics): People billed it as a [battle] for IP that's worth billions. Sure, the IP is worth billions, but that doesn't reflect what would actually be transferred. We went through that with the Alnylam story and the patent battle around RNAi.
I’m confident that ultimately this is going to be a footnote in the CRISPR story, just like it’s a footnote in the antibody story. If you look at all the antibody drugs now, does anyone remember the IP battles?
Simeon George: Everything about CRISPR has been heightened, accelerated, done in a way that is unusual. Of all the deals that I’ve worked on, this has been the most squarely in the broader ecosystem’s eye, if you will.
The lay audience has some sense of CRISPR and what's happening. This technology is very well reported across scientific publications through to the BBC, The New York Times, etc.
The company, the team, the board, investors have had to be hyper aware of all of the noise and extraneous things that are around us.
Rodger Novak (board chair, CRISPR Therapeutics): Intellia and Editas went public [before CRISPR Therapeutics], which may have helped get the message out. Today you couldn’t do this. It was a special time.
But there was also a lot of skepticism. The IPO was probably the most tiring week of my life. I would put it this way: It was so hard that in the end, when we succeeded [with the IPO], we were too tired to have a real party. We didn't even ring the bell.
Bill Lundberg: [Ethics] was another topic we talked about a fair amount. Do we have an ethical basis to continue to provide these therapies? That was a pretty straightforward conversation.
[But] there was another element: What's the rest of the world going to think of the ethics? Is the U.S. Senate suddenly going to decide that this is a terrible technology and shut it all down? Is popular culture going to blow this way out of proportion? There was a large amount of uncertainty in all of these areas.
We consulted with an ethics center at Stanford University about this. And then in a precompetitive approach, Nessan and I went to D.C. and met with the Office of Science and Technology Policy and had the opportunity to educate.
We felt we had to defend ourselves, educate, and get the message out to protect our ability to do this [research].
Nessan Bermingham (CEO, Intellia Therapeutics): The [U.S. intelligence community] put out a piece about bioterrorism and [CRISPR] as a threat. We spent a lot of time navigating that and the implications around it. It really became, for a short period of time, problematic and a threat to us. We thought about whether we’d actually be prevented from moving these technologies forward.
Sam Kulkarni: It was a wild ride. Our stock would go all the way down based on publications in academia.
But we dosed the first few patients and that was really key. In the early days, it’s hard to find these patients — new platform, new technology, unproven, etc.
Vertex Pharmaceuticals headquarters building in Boston, Massachusetts, on Sept. 21, 2017.
Bill Sikes/AP
Chapter 4 A ‘powerful partnership’ (2016 - 2021)
Venture capitalists weren’t the only ones who saw value in CRISPR. Large pharmaceutical companies moved in quickly to license the technology and partner with CRISPR Therapeutics, Intellia and Editas.
Stuart Arbuckle (Chief commercial officer, Vertex): We very early on recognized that this was going to be a generational technology and wanted to work with one of the companies. CRISPR Therapeutics was the one we selected because we thought they were the best fit for Vertex.
David Altshuler (Chief scientific officer, Vertex): It was as obvious to me as, like, where my kitchen is, that the combination of BCL11A in sickle cell and CRISPR was a possibility. When I joined Vertex, [former CEO] Jeff Leiden and I talked about what the business opportunities were. We agreed this was the right thing to do.
When we were talking to the different CRISPR companies, it was absolutely necessary to do sickle cell. We felt that that was the best opportunity.
Sam Kulkarni: The logic was, this is all new and tricky so having two parties stress test it might lead to a better product. Most biotechs don’t think about how much money they need to spend. They think about one or two years. We knew that it was going to cost over a billion dollars to develop through approval. How do we finance it all? And that was why we did a 50-50 deal with Vertex.
I think Vertex initially wanted to license the technology. They wanted to make it their own product, similar to what Biogen had done with Sangamo Biosciences. But, for us, the 50-50 partnership made a lot of sense because we had a lot of value to bring and wanted to make sure we had a lot of ownership in how the program progressed.
Nessan Bermingham: Bayer, Vertex, Regeneron, Novartis; they were playing all three [CRISPR] companies. It was basically shopping by the parties to see where they would get the best deal and where they were philosophically aligned on how to move the technology forward.
We spent a lot of time talking with Vertex. Ultimately CRISPR Therapeutics did the deal with them. In retrospect, our focus at Intellia was going in vivo and CRISPR Therapeutics was certainly more focused than we were on ex vivo applications.
Vertex Pharmaceuticals’ Chief Scientific Officer David Altshuler
Permission granted by Vertex Pharmaceuticals
David Altshuler: Bulk delivery is incredibly hard in all genetic medicines, getting the thing to where it belongs. In sickle cell, you could do ex vivo gene editing. There was so much known about bone marrow transplant and about what would happen.
Rodger Novak: A number of factors come into play when you do a partnership like that. The input cannot be just economic. There must be some kind of enabling. Vertex, at the time, was purely [a] small molecule [company]. But they had a very committed, smart team and it became clear they wanted to enter this field.
Still, it's never easy to be the junior partner, because even if we brought the knowledge, we brought the asset, you're considered by a company like Vertex as a junior partner.
Bill Lundberg: We were a small company and limited in dollars, experience and expertise. We had talked to and came very close to doing a deal with a different company around gene editing opportunities. [But] Vertex recognized the power of the human genetics that underlied the basis for this approach to sickle cell and beta thalassemia. There was a connection and appreciation there.
Bastiano Sanna (chief of cell and genetic therapies, Vertex): We, as a strategy, work only on diseases for which the underlying mechanism or cause is known. Sickle cell is one of them. It is the oldest described genetic disease in the history of medicine.
So it's really well known what the cause is. It's also known that if you increase levels of fetal hemoglobin you pretty much get rid of all the clinical consequences of sickle cell.
We chose CRISPR as the technology as opposed to, for example, viral methods, because of three reasons. One is its precision, targeting only a particular region of the genome. That of course has consequences in safety, because you know exactly what you're cutting. [The second is] efficacy, because you know the effect. [And the third is] persistence, because once those cuts are made, they are for life.
Tirtha Chakraborty: It was a powerful partnership. Vertex having been there, done that, gave a lot of power to CRISPR Therapeutics as a smaller entity that was learning how to do these things.
In 2021, after six years of working with CRISPR Therapeutics, Vertex amended their collaboration, paying $900 million to own a greater share of the profits (and the costs).
Sam Kulkarni: We realized [taking on commercialization] would have meant we'd have to hire 200 people in commercial and change how we operate. Companies like Alnylam have talked about what it meant to go commercial and how it transformed the company.
What was important to me was that we continued to be driven by research and translation as a company, and that’s what everyone spends their time on. This was the best solution. Vertex already had an established footprint and capabilities to commercialize this. They brought a lot of capital and money for CRISPR Therapeutics. They allowed us to focus on what really matters.
Victoria Gray, the first person with sickle cell disease to receive the therapy now approved as Casgevy, speaks to Haydar Frangoul, a hematologist and trial investigator
Victoria Gray
Chapter 5 Testing and results (2019 - 2023)
Early in 2019, CRISPR Therapeutics and Vertex treated the first beta thalassemia patient with Casgevy. Soon after, they treated the first sickle cell patient. The milestone came after years of preclinical research and manufacturing preparations and, while hopes were high, the outcome wasn’t certain.
Over time, early results trickled out and the companies enrolled more patients in their twin studies testing the drug in the two blood diseases.
Haydar Frangoul (hematologist, Sarah Cannon Research Institute): The preclinical data looked good. But in science, preclinical data doesn't always translate to human outcomes. So when we dosed the first patient, we were on pins and needles trying to figure out how high their fetal hemoglobin would go, and whether it would translate into clinical benefit.
Sam Kulkarni: You find a patient, then you have to collect their cells, go through manufacturing and dose them. You’re sitting on the edge of your seat for almost a six month-long journey.
You don't really know how the manufacturing goes for about two or three weeks. The key part was making sure we have the drug product manufactured. And that was probably the most nerve-wracking part of all this. Once we had the drug manufactured, the actual infusion of the patient was less climactic.
Simeon George: The biology made sense. I believed in the technology. [But] there were leaps we were taking. So there was a cautious optimism. When we first saw that clinical data though, jaws dropped. I trained as a physician and it’s literally rewriting medical textbooks. I'm not even that old. When I was in medical school, I wouldn't have imagined this.
Victoria Gray (First person with sickle cell treated with Casgevy): I didn’t want to wait. There was an urgency for me, because my life was hard. My kids began to have a fear of me dying. Their behaviors had changed in school. I knew I had to do something.
The beginning [of treatment] was still hard. I experienced body aches because of the [preparatory] chemotherapy. I didn't feel an immediate change. It was about eight months before I felt a real difference.
But within that eight months, I wasn't going to the hospital. That was new, to have an eight-month stretch without going to the emergency room or being in the hospital.
Rodger Novak: After about three months into treatment for the first patient, their fetal hemoglobin levels were so much above what I had expected. I said, ‘Oh my God, this works.’ And then I got very nervous for the next data point.
Sam Kulkarni: It was an exhilarating feeling to hear not just that the patient is doing well, but that the levels of fetal hemoglobin were remarkable. It surprised even us on the team how well it worked.
We wanted to get these remarkable data out and decided to do a company event. A lot of the company found out real time together with the rest of the world. When we finished, people had this mix of excitement, relief, joy for the patients and a feeling that we had actually built something at this company.
But it wasn't champagne glasses. We weren’t already celebrating. We kept saying to the team, ‘It’s early days. Let's just watch this.’ We needed to make sure the effects were durable. We wanted to make sure we didn’t take our eyes off the ball.
Stuart Orkin (hematologist, Boston Children’s Hospital): When they published the first paper on the data, I guess my feeling was, ‘Yeah, that's the way it should have gone. I'm glad they didn't screw it up.’ I wasn't surprised at the result, because we knew that that's what it should have been. But there was a sense of relief that it all went well.
Sam Kulkarni: The moment when I realized this was a drug was when I saw nine-month data for more than three patients. It seemed to work.
Researcher and CRISPR Therapeutics board member Katherine High
Permission granted by AskBio
Katherine High (board member, CRISPR Therapeutics): At the board level, there were probably more discussions, not about the quality of the data, but the best way to make the product available.
In the U.S., we perform about 25,000 bone marrow transplants or so per year. And there are 100,000 people with sickle cell. It's not as if those 25,000 people won't need transplants anymore. They will. So we needed to figure out how to add additional capacity.
Stuart Orkin: They did what we described in the 2015 paper. What they did — I don't want to diminish. I want to give them full credit — is the clinical execution of that, the scale-up, the quality control and the safety and all of that.
It is essential in this field because there's no tolerance for failure.
Victoria Gray: [Before] I was going to the hospital every four to six weeks to get a catheter placed in to pull out four to five units of my blood, and get replacement [blood] to keep me healthy. That was the routine.
I no longer have to do that. My blood counts remain stable. And I don’t experience pain at all from sickle cell disease.
Crescent-shaped red blood cells from a person with sickle cell disease are viewed under a microscope in 1972.
F. Gilbert/CDC/AP
Chapter 6 The first CRISPR medicine (2023)
After four years of testing, Casgevy’s benefit was clear. The treatment could eliminate the debilitating pain crises people with sickle cell frequently experience. Those with severe beta thalassemia could go without the regular blood transfusions they previously required.
The U.K.’s Medicine and Healthcare products Regulatory Agency was the first to issue a decision, clearing Casgevy for certain people with either disease 12 years or older on Nov. 16 The FDA followed on Dec. 8 and approved the therapy for people with sickle cell. Its decision in beta thalassemia is expected next year. The therapy will cost $2.2 million, Vertex said.
Sam Kulkarni: If I look back and think that, within a decade of starting a company, that we have our first approved drug, it’s just incredible.
The journey that CRISPR has been through is unlike any other. If you think of the great biotech companies right now, if you think about Alnylam or Regeneron or Vertex, they all generally took about 15 to 20 years to get their first drug approved. We’ve been able to get there much faster.
It wasn’t always a straight line. There were twists and turns that both Vertex and us navigated.
Rachel Haurwitz: To see the field actually have a first approved CRISPR-edited product plants a flag that CRISPR is here in a consequential way.
Today our world is largely two kinds of assets: small molecules and monoclonal antibodies. We are on the precipice of there actually being three legs to that stool. The third leg is genetic medicines and CRISPR is an incredibly important part of that.
Tirtha Chakraborty (chief scientific officer, Vor Biopharma): Monoclonal antibodies cannot hit anything inside the cell. Small molecules can to some extent, but they have their own limitations. CRISPR technology completely changes the world because it can get inside a cell and hit targets that were completely undruggable before. You don't need to hit a protein molecule. You can hit a part of the genome that will never be expressed in the form of a protein or RNA.
Today, we know a large part of the genome is not expressed, but plays really important roles. How do you manipulate the part that has been hiding away from all the drugs? Newer technologies exist because the previous systems have failed to address those problems.
That said, CRISPR is a classic nucleic acid therapy. Any nucleic acid therapy requires a powerful partnership with delivery technology. The success of CRISPR as a platform also brings the need for evolution.
Rodger Novak (venture partner, SR One): This goes back to academia. Thousands of labs have driven the innovation of CRISPR gene editing. You almost have the entire life sciences academic field applying a technology all together. I don’t think we’ve seen a technology democratized to this extent, except for PCR testing.
CRISPR is one of the most fundamental innovations in life science we have seen over the last 20 years. It will have a huge impact on the future.
Emmanuelle Charpentier (co-founder, CRISPR Therapeutics): This milestone certainly underscores the importance of fundamental research in the field of microbiology. I am truly amazed at the speed at which CRISPR research and applications have developed to get us to this historic moment.
My most sincere acknowledgment goes to the team at CRISPR Therapeutics for their efforts and commitment to develop the CRISPR/Cas9 technology.
Stuart Orkin: The approval is final gratification, coming full circle. I started at a time when we could barely clone genes and we had no conception whatsoever that we'd ever be able to do what we're capable of doing now. It's validation of what I've done as a career.
David Altshuler: When I was an intern at Mass General and working in the ICU, I admitted a young man who had a sickle cell crisis. He died later that day. His sickle cell had made one of his bones die, and the bone marrow went into his bloodstream. I never forgot it.
This is not a CRISPR story. CRISPR is a means to an end. The end is helping people with sickle cell.
Ben Fidler contributed reporting.
Article top image credit:
Photo illustration: Shaun Lucas/Industry Dive; CRISPR Therapeutics; Gregor Fischer/DPA/Newscom
The latest developments on the gene therapy frontier
Gene therapy has reached clinical reality with three dozen FDA approvals, but mounting safety concerns have intensified regulatory scrutiny. As developers navigate seven-figure price tags and limited adoption, the field balances breakthrough potential against sobering setbacks.
included in this trendline
UniQure says FDA wants another study of Huntington’s gene therapy
Gene therapy makers rally after news of Prasad’s exit
FDA fleshes out new roadmap for testing personalized therapies
Our Trendlines go deep on the biggest trends. These special reports, produced by our team of award-winning journalists, help business leaders understand how their industries are changing.