Bristol Myers Squibb, like many of its big pharma peers, grew to be one of the world’s largest and most powerful drugmakers via deals.
Its name stems from two 19th-century companies that merged nearly four decades ago. Its second-biggest product, the immunotherapy Opdivo, was acquired from a small, New Jersey-based developer. Its position as a leading producer of cell therapies and blood cancer treatments was forged through the landmark, $74 billion buyout of Celgene in 2019.
Now, the company is trying to find success in a new frontier. Neuroscience has long been considered one of the most challenging arenas of drug research, but Bristol Myers executives say they’re eager to establish a reputation there.
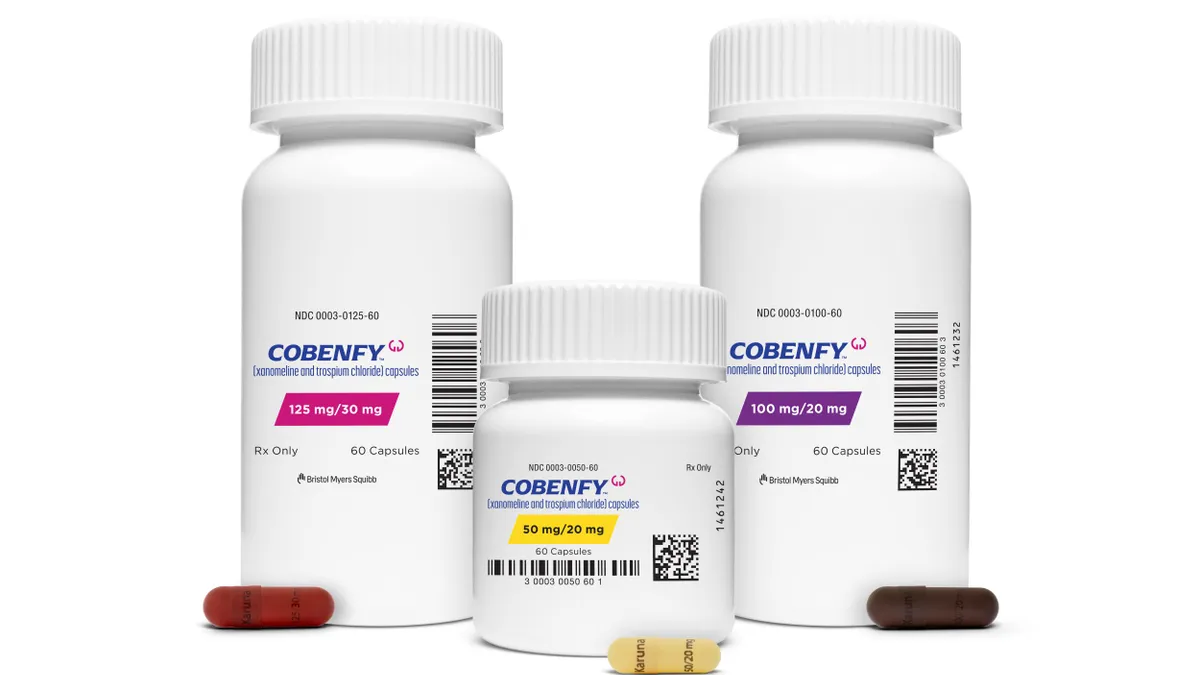
They already have a foothold, too, as a $14 billion acquisition that closed in 2024 gave the company a first-of-its-kind medicine for schizophrenia. While sales of that medicine, Cobenfy, have so far been modest, the company sees it as the backbone to a potential psychiatry franchise. Clinical trials are currently testing it as a treatment for bipolar mania as well as the psychosis and agitation associated with Alzheimer’s disease. That psychosis study is “highly anticipated by investors,” according to William Blair analyst Matt Phipps, and should produce results this year.
BioPharma Dive spoke to Ken Rhodes and Laura Gault, the respective heads of neuroscience research and development at Bristol Myers, about the kinds of mechanisms they see as worthy investments. They also offered insights about the state of Alzheimer’s and psychiatry drug research and what needs to change for those fields to propel forward.
The following conversation has been edited and condensed for clarity.
BIOPHARMA DIVE: A recently published paper stirred a lot of conversation about the amyloid hypothesis. When thinking about the spectrum of potential ways to treat Alzheimer’s, what stands out to Bristol Myers?
GAULT: We are really looking to establish a diversified and balanced pipeline across neuroscience, but also within Alzheimer's disease. We're trying to have that balance between symptomatic and disease-modifying approaches, because they certainly have different risk profiles and [vary in the] cost and time that you put into the programs.
Amyloid is the only validated target to date for Alzheimer's disease. As we think about moving into that early space before patients become symptomatic … I think it's perfectly reasonable to target amyloid at this point.
We’re already in tau. Clearly, a lot of data suggests it is intimately involved in the progression of the disease, so we're looking forward to seeing that. And because we believe in tau, we are interested in potentially building out with more compounds that would target it.
In terms of thinking about other compounds, we certainly follow the literature. There's been a lot written, probably over the last 10 to 15 years, about the role inflammation plays in Alzheimer's. We're really interested in that, and thinking about how to develop a pipeline that might address that aspect of Alzheimer's.
RHODES: The causal biology linking amyloid to Alzheimer's disease is incontrovertible. Patients who have mutations that give you more amyloid production get Alzheimer's disease at earlier ages. And we see earlier intervention gives patients a better treatment response and clearly a beneficial outcome.
There are lots of reasons to believe in amyloid-targeting therapy. It’s one of those areas in Alzheimer's disease drug development where we have fantastic tools to make sure we're enrolling patients who have amyloid pathology in our trials, and we can use amyloid imaging and now plasma-based biomarkers as a way to determine whether or not our therapies are impacting the underlying disease biology.
What's different about our tau program is it targets a region of tau most intimately associated with formation of nerve fibrillary tangles and most closely associated with the spread of tau pathology and the progression of cognitive impairment. We believe, of all the epitopes that have been tried and not been successful, we're in the region that gives us the best chance to prove the tau hypothesis.
What outside of tau and amyloid has caught your attention as a potentially worthwhile investment? For example, you have one program going after the protein “EIF2b.”
RHODES: Neuroinflammation. Pick up any recent paper on Alzheimer's disease genetic associations, you'll find the next, latest and greatest genes all impact inflammatory processes in the nervous system. Pick up any recent paper reporting biomarker linkage to Alzheimer's disease progression, you'll find markers associated with inflammatory biology.

That gives us a strong grounding in causal human biology to support going after neuroinflammation as a way to modify the course of Alzheimer's disease or provide symptomatic relief.
EIF2b is another one of those targets and pathways around the integrated stress response that has strong grounding and pathology in the brains of Alzheimer's patients. That, for us, was a good reason to go after it.
GAULT: You're not going to become a leader by being a follower. So, while amyloid is probably more derisked than some of the newer targets, we’re taking a comprehensive approach [and] are willing to move into a new area if we believe it's supported by science.
It sounds like you’re saying Bristol Myers isn’t necessarily sitting on its hands waiting for someone else to discover the next genetically validated target. You’re doing some of that work yourself and scouring the field for the best ideas.
RHODES: One thing we're also heavily invested in is making sure we have the tools in place to do a good clinical experiment and not head down any of the paths that weren’t productive in earlier Alzheimer’s disease clinical trials. Do we have the biomarker tools to know our drug is reaching the brain, that it's engaging the target biology, that we can measure? Do we have a good, rational basis for knowing we're studying the right doses?
Those later-stage Alzheimer’s experiments are usually complicated and incredibly expensive. What do you look for, other than the obvious safety and effectiveness, when deciding whether to push a molecule into more arduous studies?
RHODES: We think a lot about how to do the best experiment. We [want to] really know the mechanism, know that we're engaging it, and have a good basis for interpreting the outcome of the trial, whether it's positive or negative.
It’s crucially important to have all the tools in place so that, when we hand candidates over to Laura’s team, they have what they need to actually do that good experiment. We focus a lot on: How do we get as early a read as possible on whether we're on the right track?
GAULT: Taking a molecule that hasn't been in humans and going to [proof-of-concept testing], that's a huge step. There are smaller steps we can take before we get to that big, expensive study: showing the drug gets into the brain, showing the drug is engaging the target, showing that when that target is engaged, it's doing what we would expect it to do based on how we understand the biology.
As you go through development, and you look at each of these steps, you find reassuring pieces of evidence. Each of those is increasing your probability of success a bit. Each of those is increasing our willingness to invest further in the program.
There have been debates about a potential efficacy ceiling for amyloid antibodies. How are you thinking about the next disease-modifying Alzheimer’s drug? Is it a more powerful anti-amyloid therapy, or is it something that pairs really well with other drugs? How are you going to beat what’s available?
GAULT: The easy answer to your question is yes, both.
To say that we’ve reached the ceiling on amyloid and what it can deliver when you target it, I think is not correct for a couple of reasons. One is, the way that we've looked at the impact of these anti-amyloid antibodies is by looking over relatively short periods – 18 months. When you continue to follow people, over time the difference between placebo and drug widens.
So, in a way, what we see in the clinical trials is just the beginning of the effect.
It's also the case that the current medications are what we call naked antibodies, so they don't have anything attached that helps them easily get across the blood-brain barrier. In those circumstances, a very small amount actually crosses, probably 0.5%. There's a whole generation of antibodies in development, trontinemab from Roche being in the lead, that are attaching these anti-amyloid antibodies to a transporter [so they can cross this] barrier and increase levels.
When those are up and running, I think it's reasonably likely to see we haven't reached an efficacy ceiling.
Do we think that an anti-amyloid agent is going to be the full answer to Alzheimer's? That is an interesting question. I would say, once somebody is symptomatic, probably not.
As you get later in the course of the disease and patients are having symptoms, that's when you see the other pathologies coming into play. You've got a lot of inflammation at that point. You have tau accumulation. Expecting that an amyloid-lowering drug alone is going to cure the disease or stop the progression is probably not reasonable. You see the field believes that generally, because they're taking additional bets on tau and in other areas.
RHODES: What we're seeing now, with the advent of these shuttle technologies, is much more antibody in the brain. Molecules are entering through the capillary network, instead of through larger vessels where there tends to be a lot of amyloid.
The benefit-risk profile — in terms of clearing amyloid from the brain without some of the side effects that have been associated with earlier anti-amyloid therapies — seems to be greater. It may let us push to higher doses. It may let us more rapidly remove amyloid plaque without some of those risks.
The other point, which is, in many ways, the most important one, is when we look at how we're using anti-amyloids today, we're treating Alzheimer's patients [who are] almost at the maximum of amyloid accumulation.
We can always ask the question: is that just too late to really get the maximum benefit from an amyloid-targeting therapy? I firmly believe if we can walk back the initiation of treatment to much earlier … then you can start to realize the maximum benefit. How much? I don't think we've seen the ceiling yet.
Early testing then becomes vital. But that still remains challenging. How do you factor such obstacles into your decisions? Until testing gets better, does it make sense to throw a bunch of amyloid programs at the wall hoping any of them stick?

RHODES: The first question we have to answer is: How do we identify those pre-symptomatic patients at risk for developing disease, and can we get a sense of how quickly they might progress? This is where all the biomarker discovery work happening across the Alzheimer's disease space … is so critical to the future of developing therapies.
Just imagine a future where you go to your doctor for an annual checkup and get a panel of plasma biomarkers that not only tells you your risk for diabetes or cardiovascular disease, but gives you a risk score for Alzheimer's disease. Using those biomarkers, you could decide with your doctor whether to start you on a potential therapy that could prevent or delay the time to which you might develop the disease. Incredible investments are being made in biomarker discovery.
And they are going to be absolutely essential, because I don't think we can run a 10-year clinical study in patients if we don't know their risk of disease progression. That would be an extremely large and expensive trial to run. But I don't think those types of studies are too far off, given the way the biomarker field is evolving. I'm pretty optimistic.
We're already seeing some of our peer companies starting to try and run trials in this very early population. I think we'll get there.
One area that has even fewer biomarkers than Alzheimer’s is psychiatry, which you invested heavily in by acquiring Cobenfy. How are you viewing the translational research aspect of psychiatric disorders?
GAULT: In psychiatry, I can't point to a large dataset that's available to help us understand a biomarker approach. For BMS and for other companies interested in this space, we're going to need to collaborate in a pre-competitive way to establish those datasets to help guide decision making.
That said, there are some biomarkers in the psychiatry space that can be used today. EEG is an example where you can look preclinically, in animal models, and see changes in the brain circuitry that are elicited by administration of a drug. Then you look in people to see if there are corresponding changes. It helps you understand if you're getting the drug into the brain, and if you're engaging the downstream target in the way you would like to.
EEG is something used already by some companies to be successful. We're certainly thinking about that for elements of our portfolio. There are other things — functional MRI, resting state MRI — that may give some view into the circuitry of the brain and how it's operating and how your drug changes things.
RHODES: This initiative in the pre-competitive space … is going to be extremely valuable. It's early, though I think the correlations they're trying to draw between blood-based markers, cerebrospinal fluid markers, EEG and other endpoints that could help with the translational piece are going to prove to be extremely valuable.
Cobenfy-related programs account for about half of your neuroscience pipeline. On Wall Street, there have been concerns about the drug’s commercial viability. Has Cobenfy’s performance caused you to change or tailor your interest in psychiatry and what drugs you’re willing to invest in?
GAULT: It hasn't changed at all. We have a high degree of confidence in Cobenfy and what it can deliver. We're excited to continue to build that breadth in our pipeline in psychiatry and neurology moving forward.
We'll learn from Cobenfy as these trials read out. But at this point, there is no reason not to be confident that the data will be positive.